

Single-cell multi-omics profiling uncovers the immune heterogeneity in HIV-infected immunological non-responders
- PMID: 40184908
- PMCID: PMC12002939
- DOI: 10.1016/j.ebiom.2025.105667
Single-cell multi-omics profiling uncovers the immune heterogeneity in HIV-infected immunological non-responders
Abstract
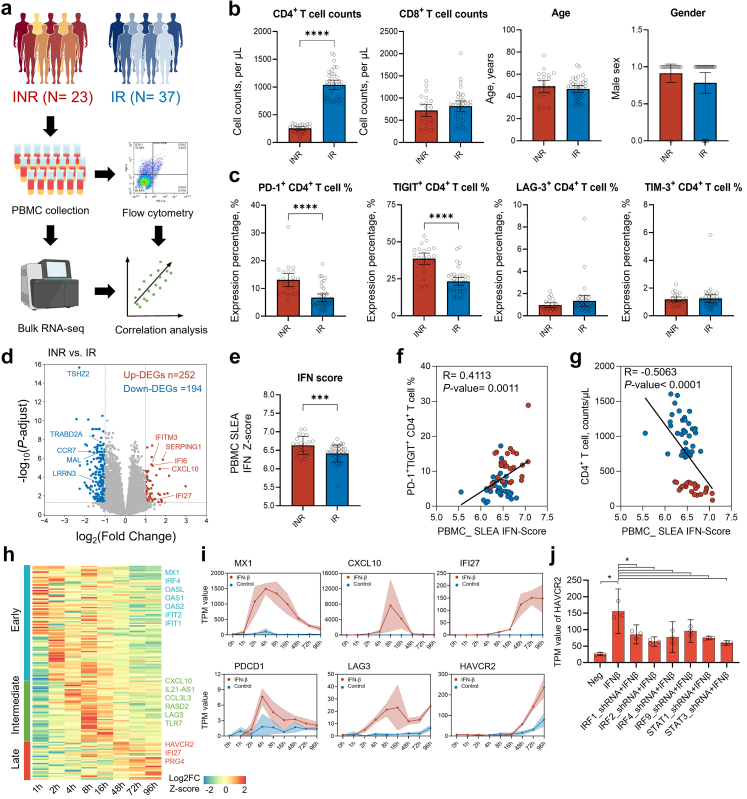
Background: Immunological non-responders (INRs) are people living with HIV-1 who fail to achieve full immune reconstitution despite long-term effective antiretroviral therapy (ART). This incomplete recovery of CD4+ T cells increase the risk of opportunistic infections and non-AIDS-related morbidity and mortality. Understanding the mechanisms driving this immune dysfunction is critical for developing targeted therapies.
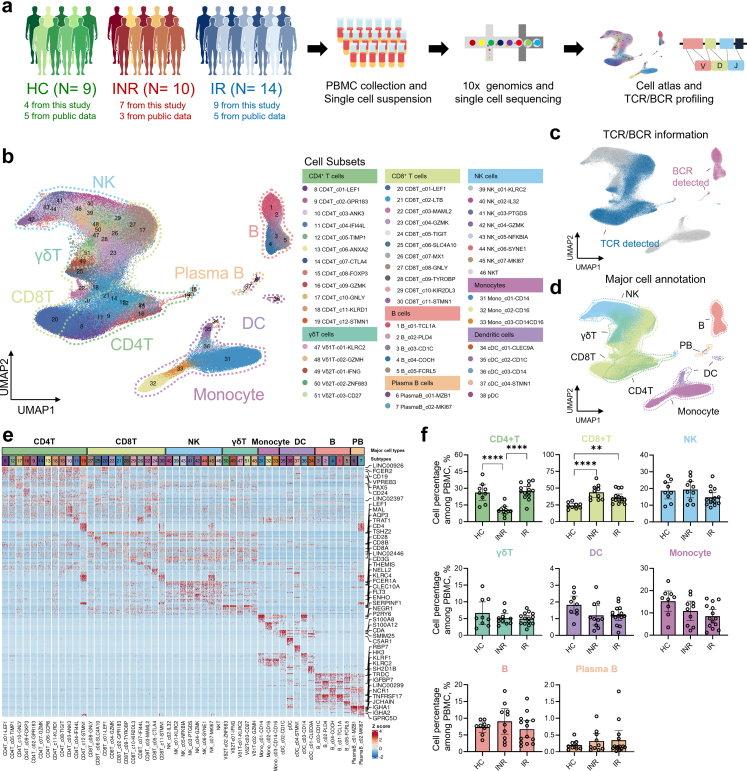
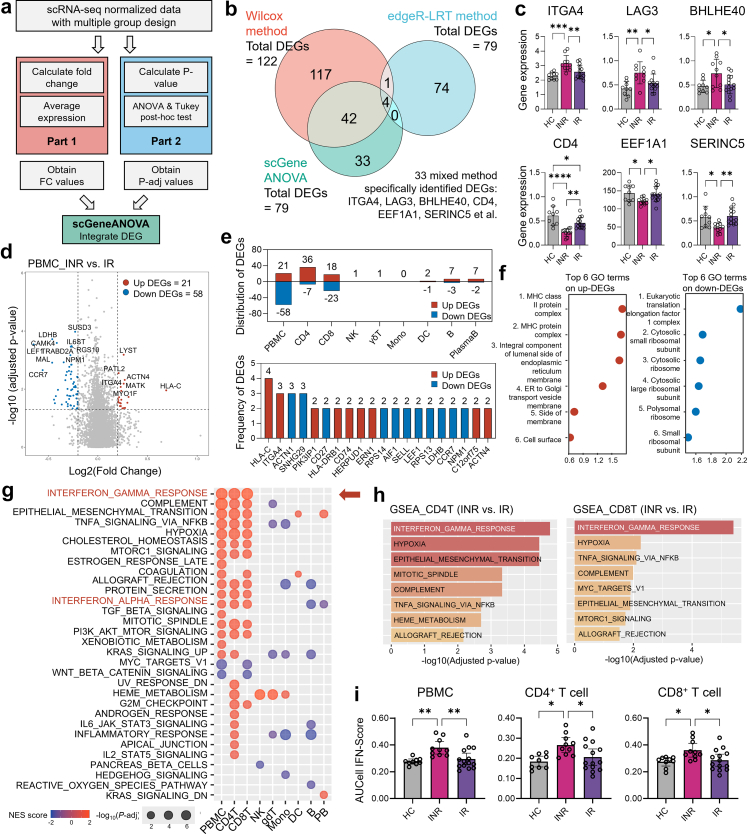
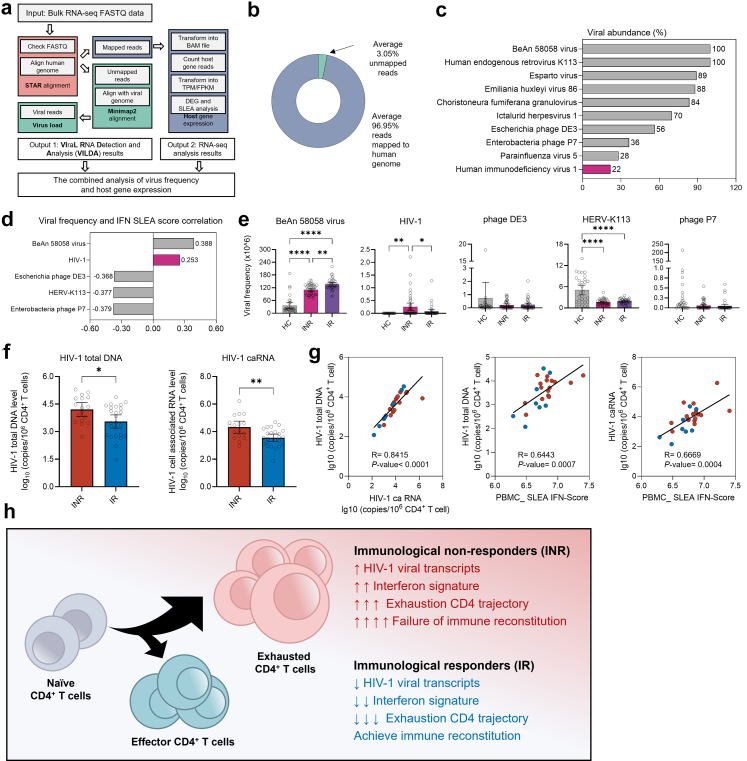
Methods: We performed single-cell RNA sequencing (scRNA-seq) and single-cell VDJ sequencing (scVDJ-seq) on peripheral blood mononuclear cells (PBMCs) from INRs, immune responders (IRs), and healthy controls (HCs). We developed scGeneANOVA, a novel mixed model differential gene analysis tool, to detect differentially expressed genes and pathways. In addition, we developed the Viral Identification and Load Detection Analysis (VILDA) tool to quantify HIV-1 transcripts and investigate their relationship with interferon (IFN) pathway activation.
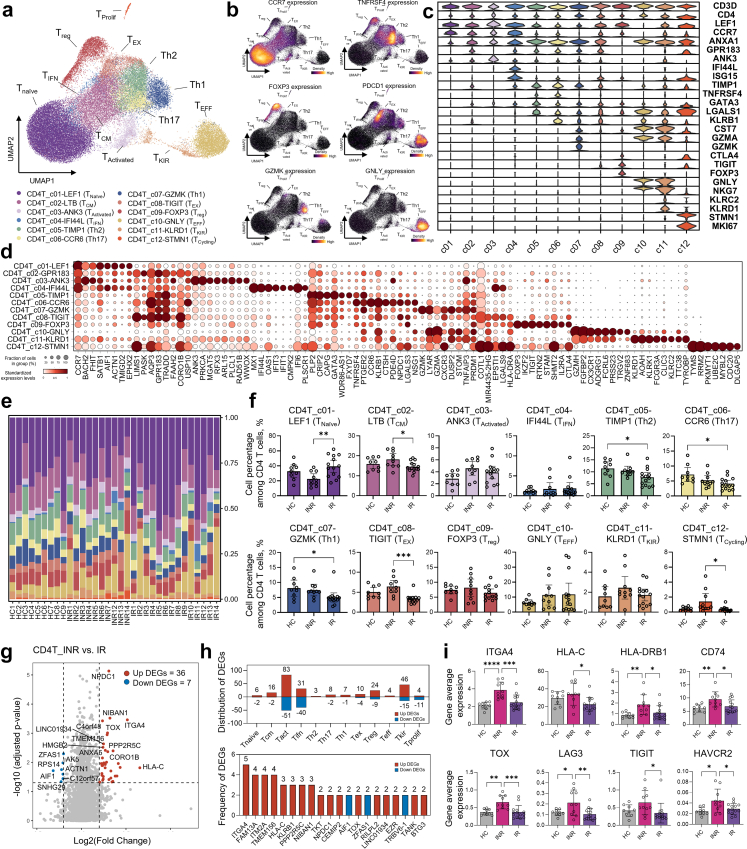
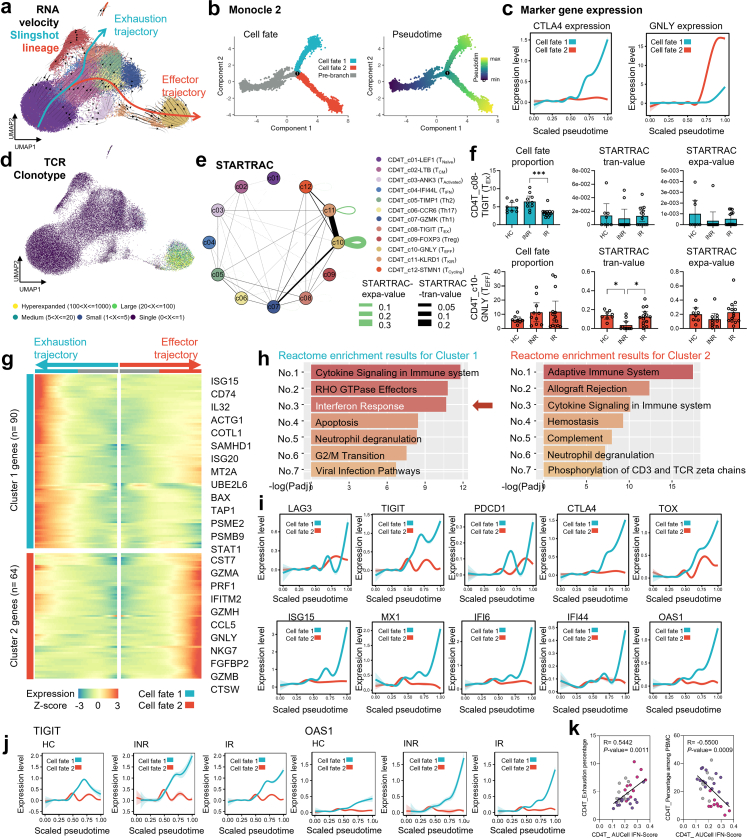
Findings: Our analysis revealed that INRs exhibit a dysregulated IFN response, closely associated with CD4+ T cell exhaustion and immune recovery failure. The scGeneANOVA tool identified critical genes and pathways that were missed by traditional analysis methods, while VILDA showed higher levels of HIV-1 transcripts in INRs, which may drive the heightened IFN response. These findings support a potential contribution of IFN signalling in INR-related immune dysfunction.
Interpretation: Our study provides new insights into the pathogenic mechanisms behind immune recovery failure in INRs, suggesting that IFN signalling might be involved in the development of CD4+ T cell exhaustion. The identification of key genes and pathways offers potential biomarkers and therapeutic targets for improving immune recovery in this vulnerable population.
Funding: This study was supported by the grants from Special Research Fund for the Central High-level Hospitals of Peking Union Medical College Hospital (Grant No. 2022-PUMCH-D-008), Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Sciences (Grant No. 2021-I2M-1-037), National Key Technologies R&D Program for the 13th Five-year Plan (Grant No. 2017ZX10202101-001). The funders played no role in the design, experiment conduction, data analysis and preparation of the manuscript of this work.
Keywords: Acquired immunodeficiency syndrome; CD4(+) T cells; Human immunodeficiency virus-1; Immunological non-responders; Interferon; Single cell RNA sequencing.
Copyright © 2025 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of interests The authors have declared that no conflict of interest exists.
Figures
Similar articles
-
Multi-omics dissection of metabolic dysregulation associated with immune recovery in people living with HIV-1.J Transl Med. 2025 Jan 31;23(1):143. doi: 10.1186/s12967-025-06168-0. J Transl Med. 2025. PMID: 39891216 Free PMC article.
-
Impaired immune reconstitution in HIV infection: the role of CD4+ T-cell-associated NKG2D ligands, CD4+ T-cell subsets imbalance, and immune function deficiency.Front Immunol. 2025 Feb 21;16:1541574. doi: 10.3389/fimmu.2025.1541574. eCollection 2025. Front Immunol. 2025. PMID: 40061947 Free PMC article.
-
Comparative Transcriptional Analysis Identified Characteristic Genes and Patterns in HIV-Infected Immunological Non-Responders.Front Immunol. 2022 Jan 28;13:807890. doi: 10.3389/fimmu.2022.807890. eCollection 2022. Front Immunol. 2022. PMID: 35154126 Free PMC article.
-
Advances in Mechanism of HIV-1 Immune Reconstitution Failure: Understanding Lymphocyte Subpopulations and Interventions for Immunological Nonresponders.J Immunol. 2024 Jun 1;212(11):1609-1620. doi: 10.4049/jimmunol.2300777. J Immunol. 2024. PMID: 38768409 Review.
-
Multi-omics in HIV: searching insights to understand immunological non-response in PLHIV.Front Immunol. 2023 Aug 15;14:1228795. doi: 10.3389/fimmu.2023.1228795. eCollection 2023. Front Immunol. 2023. PMID: 37649488 Free PMC article. Review.
References
-
- Maartens G., Celum C., Lewin S.R. HIV infection: epidemiology, pathogenesis, treatment, and prevention. Lancet. 2014;384(9939):258–271. - PubMed
-
- Li T.S., Tubiana R., Katlama C., Calvez V., Ait Mohand H., Autran B. Long-lasting recovery in CD4 T-cell function and viral-load reduction after highly active antiretroviral therapy in advanced HIV-1 disease. Lancet. 1998;351(9117):1682–1686. - PubMed
-
- Autran B., Carcelain G., Li T.S., et al. Positive effects of combined antiretroviral therapy on CD4+ T cell homeostasis and function in advanced HIV disease. Science. 1997;277(5322):112–116. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
