

Severus detects somatic structural variation and complex rearrangements in cancer genomes using long-read sequencing
- PMID: 40185952
- PMCID: PMC12483193
- DOI: 10.1038/s41587-025-02618-8
Severus detects somatic structural variation and complex rearrangements in cancer genomes using long-read sequencing
Abstract
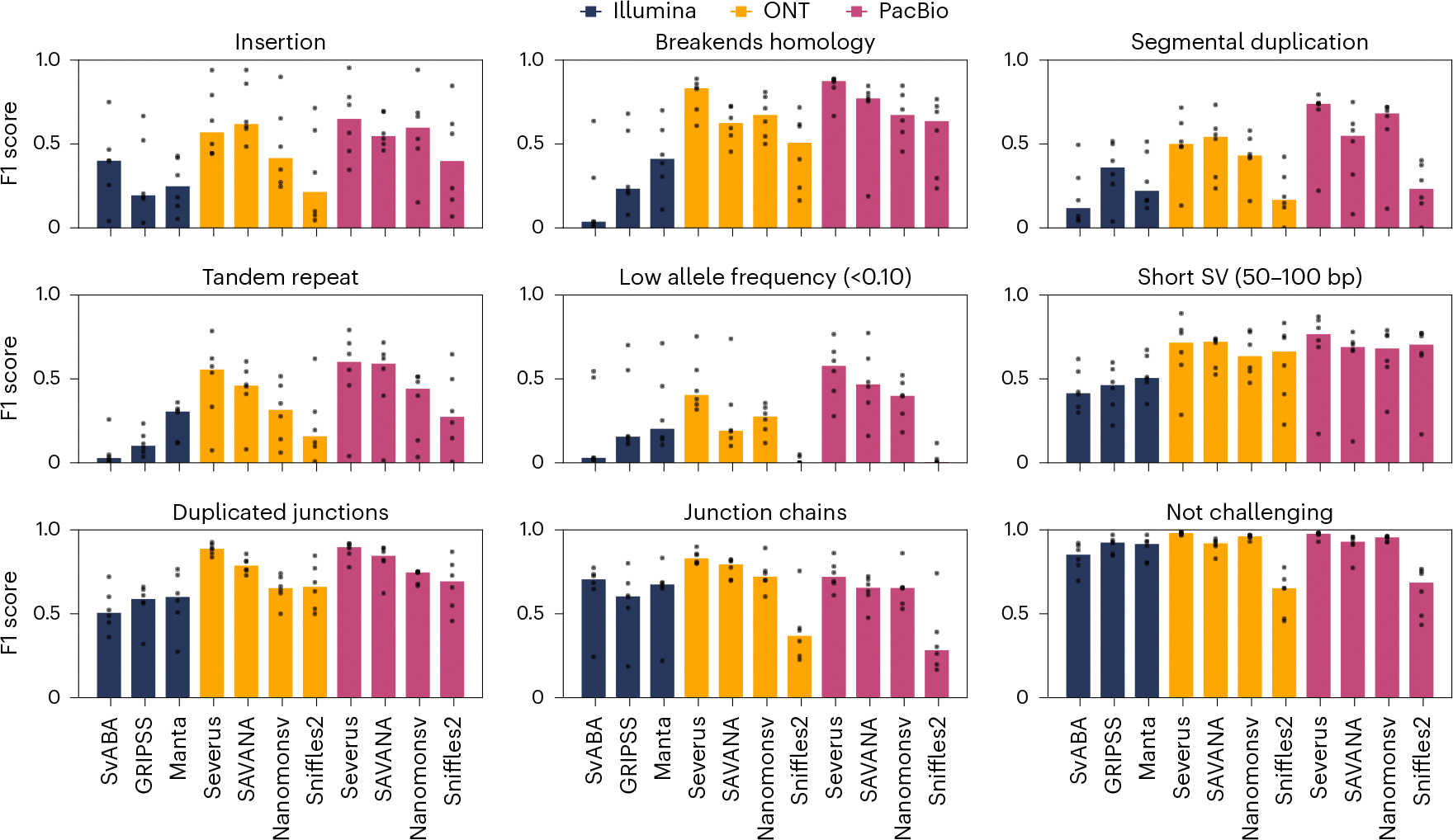
For the detection of somatic structural variation (SV) in cancer genomes, long-read sequencing is advantageous over short-read sequencing with respect to mappability and variant phasing. However, most current long-read SV detection methods are not developed for the analysis of tumor genomes characterized by complex rearrangements and heterogeneity. Here, we present Severus, a breakpoint graph-based algorithm for somatic SV calling from long-read cancer sequencing. Severus works with matching normal samples, supports unbalanced cancer karyotypes, can characterize complex multibreak SV patterns and produces haplotype-specific calls. On a comprehensive multitechnology cell line panel, Severus consistently outperforms other long-read and short-read methods in terms of SV detection F1 score (harmonic mean of the precision and recall). We also illustrate that compared to long-read methods, short-read sequencing systematically misses certain classes of somatic SVs, such as insertions or clustered rearrangements. We apply Severus to several clinical cases of pediatric leukemia/lymphoma, revealing clinically relevant cryptic rearrangements missed by standard genomic panels.
© 2025. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
Competing interests: S.A. is an employee and stockholder of ONT. A.K., P.C., K.S., D.C. and A.C. are employees of Google and own Alphabet stock as part of the standard compensation package. E.G. served on advisory boards for Jazz Pharmaceuticals and Syndax Pharmaceuticals. M.S.F. is part of the speakers bureau for Bayer and PacBio. The other authors declare no competing interests.
Figures















References
-
- Cosenza MR, Rodriguez-Martin B & Korbel JO Structural variation in cancer: role, prevalence, and mechanisms. Annu. Rev. Genomics Hum. Genet. 23, 123–152 (2022). - PubMed
MeSH terms
Grants and funding
- R01HG011274/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
- Intramural Research Program of the Center for Cancer Research/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- OT2 OD033761/OD/NIH HHS/United States
- U41HG010972/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
- Intramural funding/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- U24 HG010262/HG/NHGRI NIH HHS/United States
- U01HG013748/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
- R01 HG010485/HG/NHGRI NIH HHS/United States
- U01CA253405/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- U24HG010262/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
- R01HG010485/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
- U41 HG010972/HG/NHGRI NIH HHS/United States
- OT2OD033761/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- U01 CA253405/CA/NCI NIH HHS/United States
- R01 HG011274/HG/NHGRI NIH HHS/United States
- U24HG011853/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
- U24 HG011853/HG/NHGRI NIH HHS/United States
- U01 HG013748/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
