

Multi-targeted, NOT gated CAR-T cells as a strategy to protect normal lineages for blood cancer therapy
- PMID: 40191207
- PMCID: PMC11968376
- DOI: 10.3389/fimmu.2025.1493329
Multi-targeted, NOT gated CAR-T cells as a strategy to protect normal lineages for blood cancer therapy
Abstract
Introduction: Despite advances in treatment of blood cancers, several-including acute myeloid leukemia (AML)-continue to be recalcitrant. Cell therapies based on chimeric antigen receptors (CARs) have emerged as promising approaches for blood cancers. However, current CAR-T treatments suffer from on-target, off-tumor toxicity, because most familiar blood cancer targets are also expressed in normal lineages. In addition, they face the common problem of relapse due to target-antigen loss. Cell therapeutics engineered to integrate more than one signal, often called logic-gated cells, can in principle achieve greater selectivity for tumors.
Methods: We applied such a technology, a NOT gated system called Tmod™ that is being developed to treat solid-tumor patients, to the problem of therapeutic selectivity for blood cancer cells.
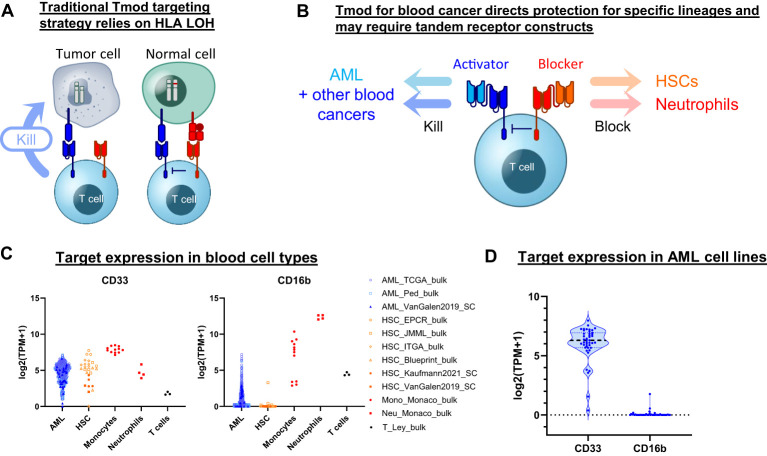
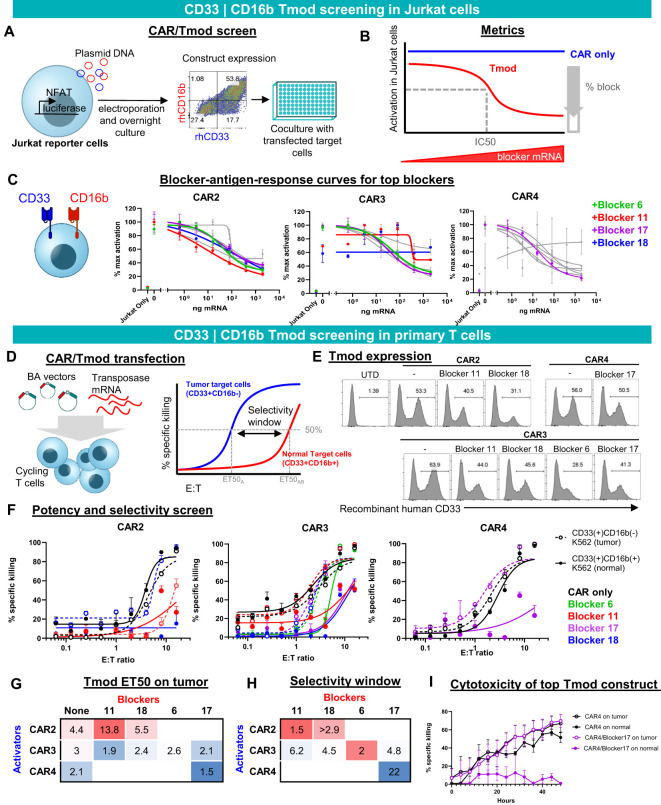
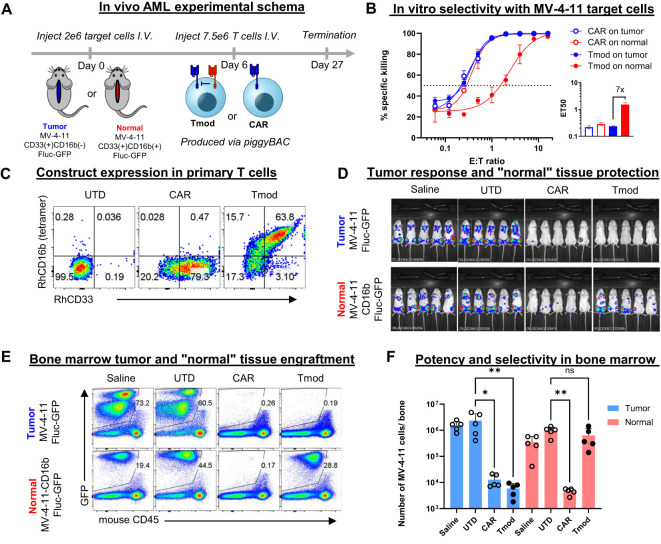
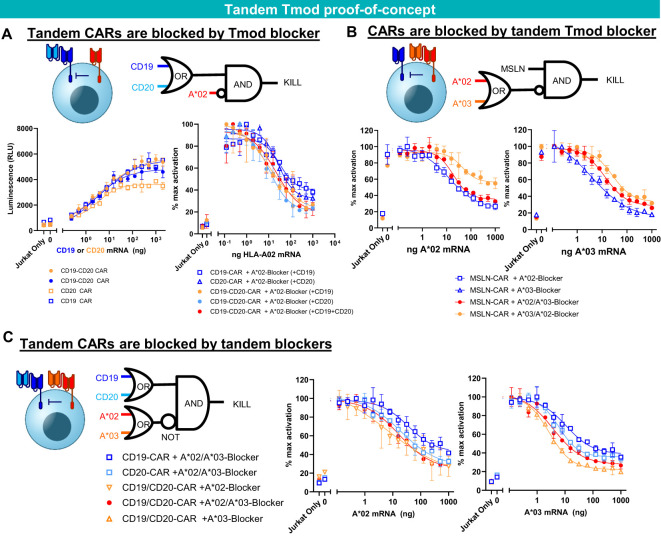
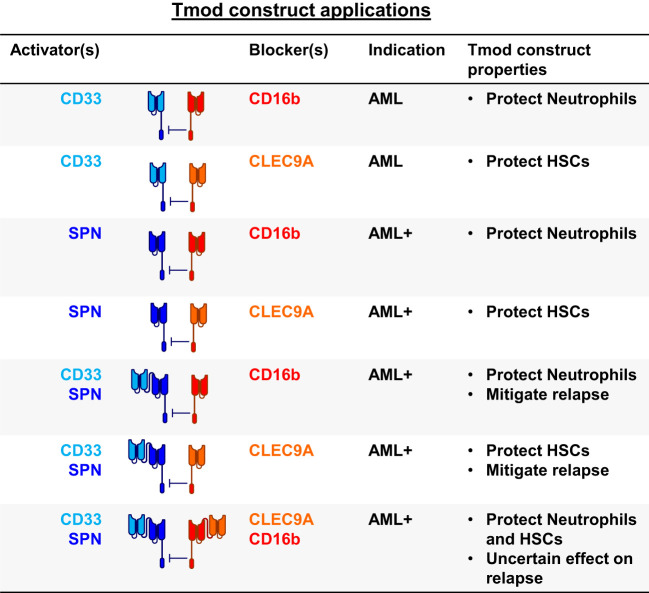
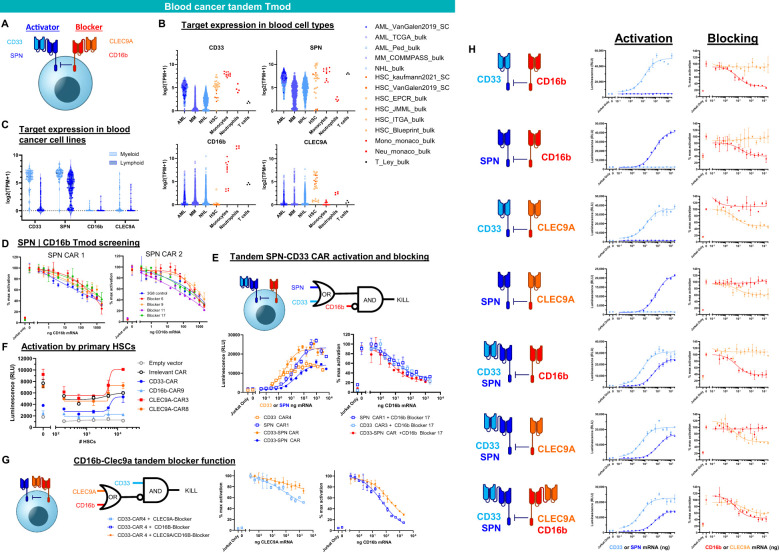
Results: Here we show that Tmod cells can be designed to target 2-4 antigens to provide different practical and conceptual options for a blood cancer therapy: (i) mono- and bispecific activating receptors that target CD33, a well-known AML antigen expressed on the majority of AML tumors (as well as healthy myeloid cells) and CD43 (SPN), an antigen expressed on many hematopoietic cancers (and normal blood lineages); and (ii) mono- and bispecific inhibitory receptors that target CD16b (FCGR3B) and CLEC9A, antigens expressed on key normal blood cells but not on most blood cancers.
Discussion: These results further demonstrate the robust modularity of the Tmod system and generalize the Tmod approach beyond solid tumors.
Keywords: AML; CAR-T cells; CD16; CD33; LIR-1 (LILRB1); SPN; blood cancer; logic gate.
Copyright © 2025 DiAndreth, Nesterenko, Winters, Flynn, Jette, Suryawanshi, Shafaattalab, Martire, Daris, Moore, Elshimali, Gill, Riley, Miller, Netirojjanakul, Hamburger and Kamb.
Conflict of interest statement
All authors are current or former employees and shareholders of A2 Biotherapeutics, Inc.
Figures
References
-
- Mardiros A, Dos Santos C, McDonald T, Brown CE, Wang X, Budde LE, et al. . T cells expressing CD123-specific chimeric antigen receptors exhibit. specific cytolytic effector functions antitumor effects against Hum acute myeloid leukemia. Blood. (2013) 122:3138–48. doi: 10.1182/blood-2012-12-474056 - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical