

This is a preprint.
Combining multiplexed functional data to improve variant classification
- PMID: 40196145
- PMCID: PMC11975307
Combining multiplexed functional data to improve variant classification
Abstract
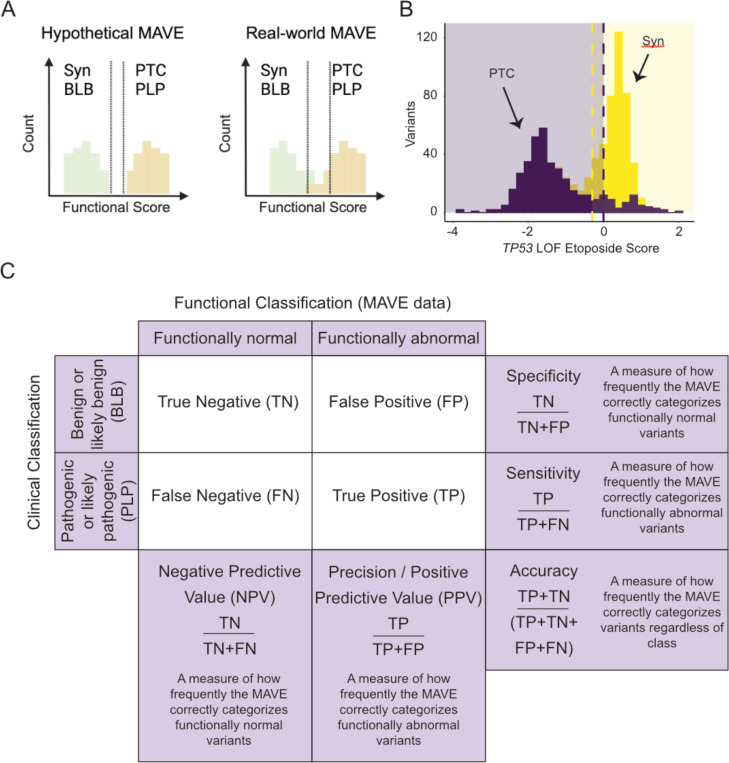
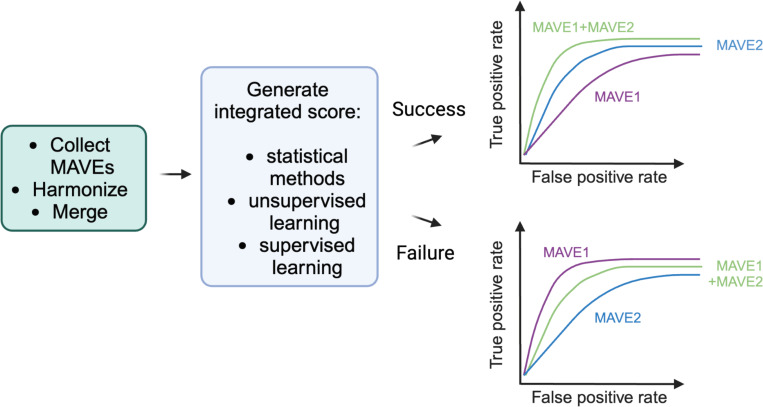
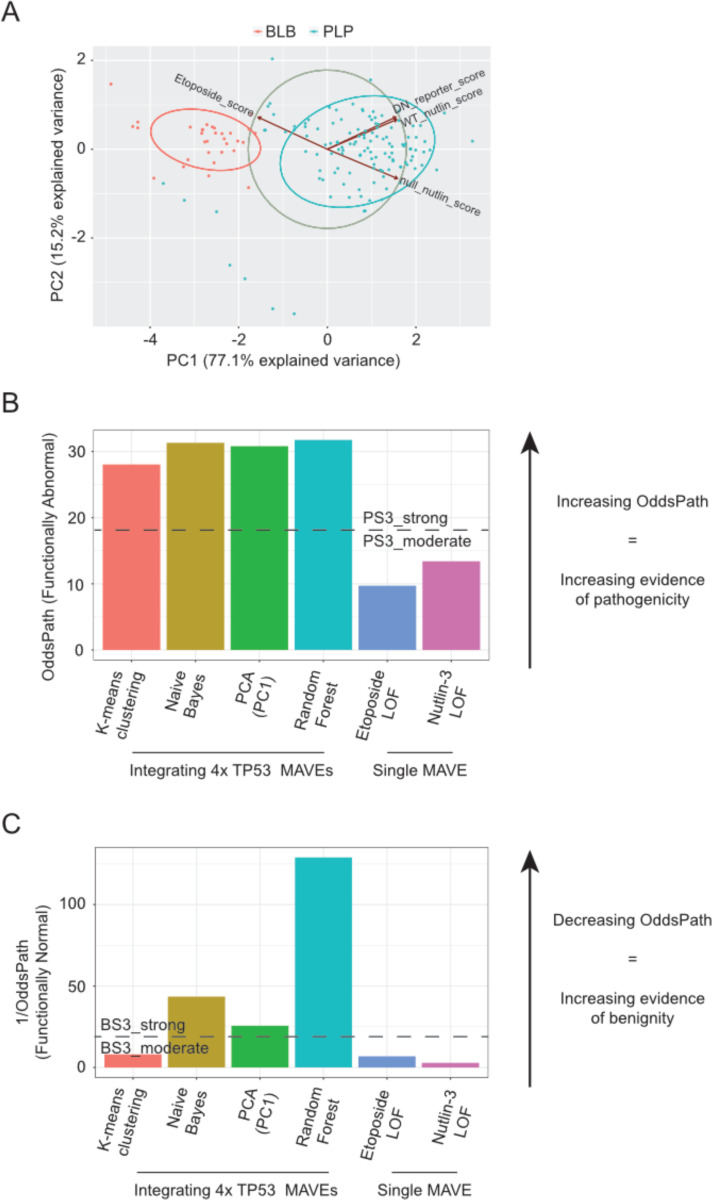
With the surge in the number of variants of uncertain significance (VUS) reported in ClinVar in recent years, there is an imperative to resolve VUS at scale. Multiplexed assays of variant effect (MAVEs), which allow the functional consequence of 100s to 1000s of genetic variants to be measured in a single experiment, are emerging as a source of evidence which can be used for clinical gene variant classification. Increasingly, there are multiple published MAVEs for the same gene, sometimes measuring different aspects of variant impact. Where multiple functional consequences may need to be considered to get a more complete understanding of variant effects for a given gene, combining data from multiple MAVEs may lead to the assignment of increased evidence strength which could impact variant classifications. Here, we provide guidance for combining such multiplexed functional data, incorporating a stepwise process from data curation and collection to model generation and validation. We illustrate the potential of this approach by showing the integration of multiplexed functional data from four MAVEs for the gene TP53. By following these steps, researchers can maximize the value of MAVEs, strengthen the functional evidence for clinical variant classification, reclassify more VUS, and potentially uncover novel mechanisms of pathogenicity for clinically relevant genes.
Figures
Similar articles
-
Multimodal framework to resolve variants of uncertain significance in TSC2.bioRxiv [Preprint]. 2024 Jun 8:2024.06.07.597916. doi: 10.1101/2024.06.07.597916. bioRxiv. 2024. PMID: 38895336 Free PMC article. Preprint.
-
Using multiplexed functional data to reduce variant classification inequities in underrepresented populations.Genome Med. 2024 Dec 3;16(1):143. doi: 10.1186/s13073-024-01392-7. Genome Med. 2024. PMID: 39627863 Free PMC article.
-
Closing the gap: Systematic integration of multiplexed functional data resolves variants of uncertain significance in BRCA1, TP53, and PTEN.Am J Hum Genet. 2021 Dec 2;108(12):2248-2258. doi: 10.1016/j.ajhg.2021.11.001. Epub 2021 Nov 17. Am J Hum Genet. 2021. PMID: 34793697 Free PMC article.
-
Assigning credit where it's due: An information content score to capture the clinical value of Multiplexed Assays of Variant Effect.bioRxiv [Preprint]. 2023 Oct 20:2023.10.20.562794. doi: 10.1101/2023.10.20.562794. bioRxiv. 2023. Update in: BMC Bioinformatics. 2024 Sep 6;25(1):295. doi: 10.1186/s12859-024-05920-5. PMID: 37905042 Free PMC article. Updated. Preprint.
-
Navigating variants of uncertain significance in genetic dyslipidemia: how to assess and counsel patients.Curr Opin Lipidol. 2025 Apr 1;36(2):49-54. doi: 10.1097/MOL.0000000000000971. Epub 2024 Dec 18. Curr Opin Lipidol. 2025. PMID: 39950242 Review.
References
-
- Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, Karapetyan K, Katz K, Liu C, Maddipatla Z, Malheiro A, McDaniel K, Ovetsky M, Riley G, Zhou G, Holmes JB, Kattman BL, Maglott DR. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062–D7. doi: 10.1093/nar/gkx1153.. - DOI - PMC - PubMed
-
- Dawood M, Fayer S, Pendyala S, Post M, Kalra D, Patterson K, Venner E, Muffley LA, Fowler DM, Rubin AF, Posey JE, Plon SE, Lupski JR, Gibbs RA, Starita LM, Robles-Espinoza CD, Coyote-Maestas W, Gallego Romero I. Defining and Reducing Variant Classification Disparities. medRxiv. 2024. Epub . doi: 10.1101/2024.04.11.24305690.. - DOI - PMC - PubMed
-
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. Epub 20150305. doi: 10.1038/gim.2015.30.. - DOI - PMC - PubMed
-
- Buckley M, Terwagne C, Ganner A, Cubitt L, Brewer R, Kim DK, Kajba CM, Forrester N, Dace P, De Jonghe J, Shepherd STC, Sawyer C, McEwen M, Diederichs S, Neumann-Haefelin E, Turajlic S, Ivakine EA, Findlay GM. Saturation genome editing maps the functional spectrum of pathogenic VHL alleles. Nat Genet. 2024;56(7):1446–55. Epub 20240705. doi: 10.1038/s41588-024-01800-z.. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous