

MSFragger-DDA+ enhances peptide identification sensitivity with full isolation window search
- PMID: 40199897
- PMCID: PMC11978857
- DOI: 10.1038/s41467-025-58728-z
MSFragger-DDA+ enhances peptide identification sensitivity with full isolation window search
Abstract
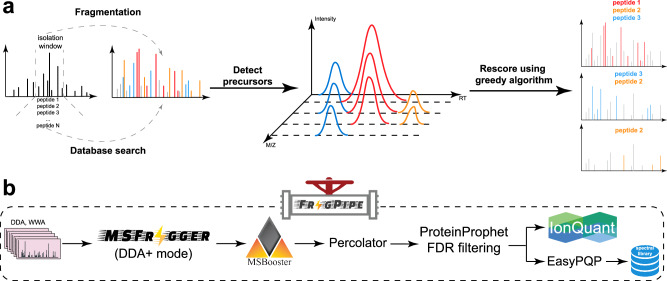
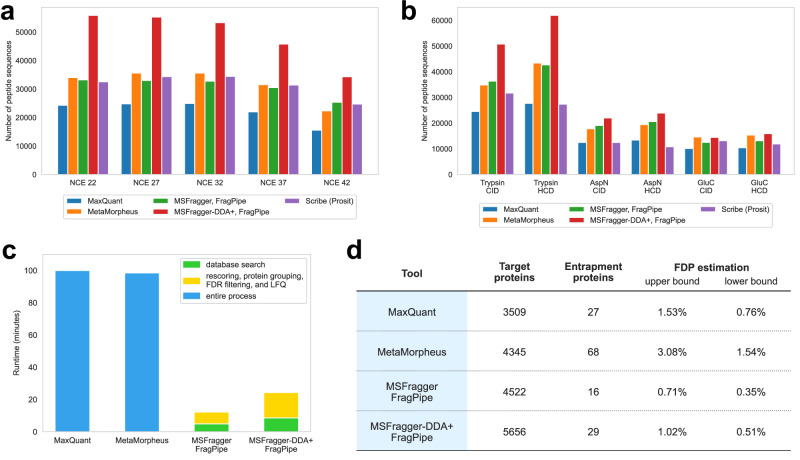
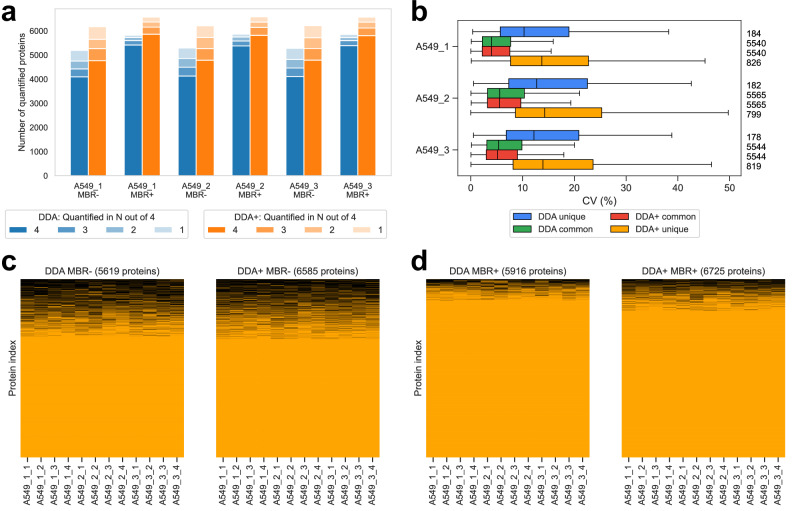
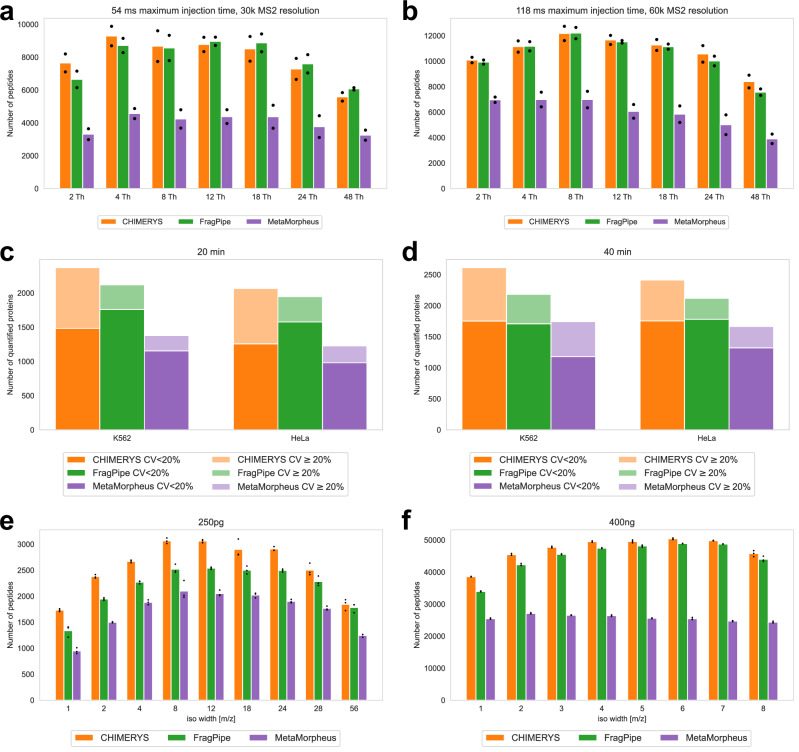
Liquid chromatography-mass spectrometry based proteomics, particularly in the bottom-up approach, relies on the digestion of proteins into peptides for subsequent separation and analysis. The most prevalent method for identifying peptides from data-dependent acquisition mass spectrometry data is database search. Traditional tools typically focus on identifying a single peptide per tandem mass spectrum, often neglecting the frequent occurrence of peptide co-fragmentations leading to chimeric spectra. Here, we introduce MSFragger-DDA+, a database search algorithm that enhances peptide identification by detecting co-fragmented peptides with high sensitivity and speed. Utilizing MSFragger's fragment ion indexing algorithm, MSFragger-DDA+ performs a comprehensive search within the full isolation window for each tandem mass spectrum, followed by robust feature detection, filtering, and rescoring procedures to refine search results. Evaluation against established tools across diverse datasets demonstrated that, integrated within the FragPipe computational platform, MSFragger-DDA+ significantly increases identification sensitivity while maintaining stringent false discovery rate control. It is also uniquely suited for wide-window acquisition data. MSFragger-DDA+ provides an efficient and accurate solution for peptide identification, enhancing the detection of low-abundance co-fragmented peptides. Coupled with the FragPipe platform, MSFragger-DDA+ enables more comprehensive and accurate analysis of proteomics data.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: A.I.N. and F.Y. receive royalties from the University of Michigan for the sale of MSFragger, IonQuant, and diaTracer software licenses to commercial entities. All license transactions are managed by the University of Michigan Innovation Partnerships office, and all proceeds are subject to university technology transfer policy. Other authors declare no competing interests.
Figures
Update of
-
MSFragger-DDA+ Enhances Peptide Identification Sensitivity with Full Isolation Window Search.bioRxiv [Preprint]. 2024 Oct 15:2024.10.12.618041. doi: 10.1101/2024.10.12.618041. bioRxiv. 2024. Update in: Nat Commun. 2025 Apr 8;16(1):3329. doi: 10.1038/s41467-025-58728-z. PMID: 39463976 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
