

Computation of Protein-Ligand Binding Free Energies with a Quantum Mechanics-Based Mining Minima Algorithm
- PMID: 40202178
- PMCID: PMC12020368
- DOI: 10.1021/acs.jctc.4c01707
Computation of Protein-Ligand Binding Free Energies with a Quantum Mechanics-Based Mining Minima Algorithm
Abstract
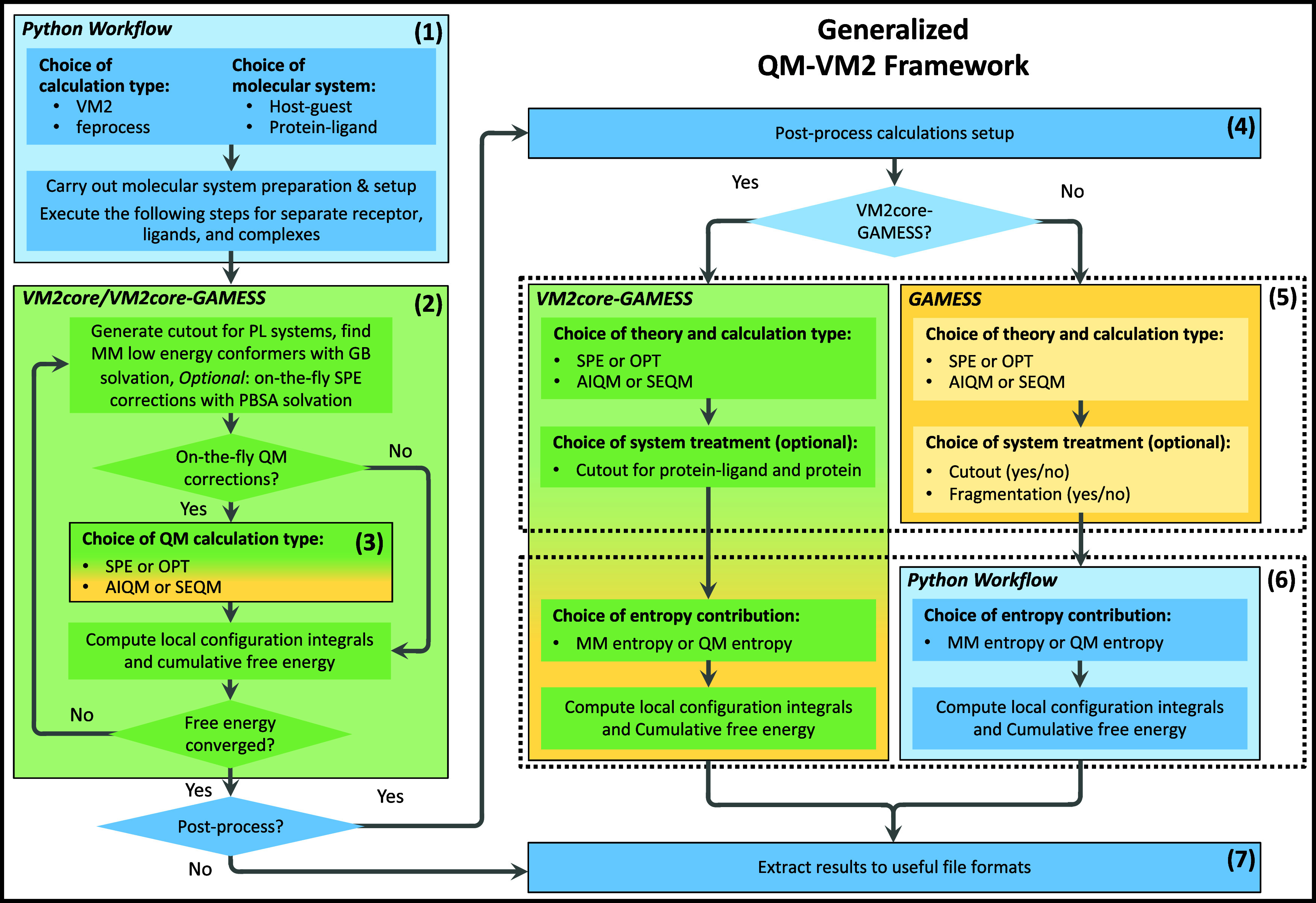
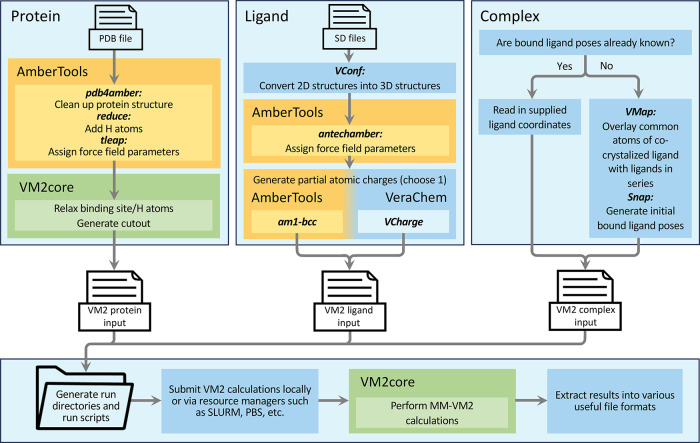
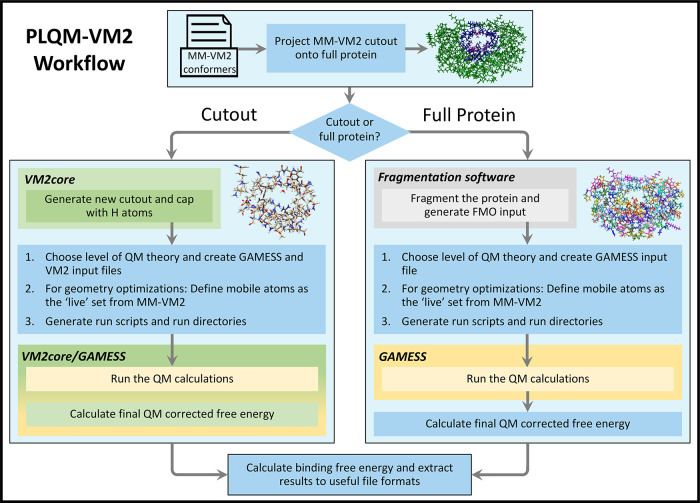
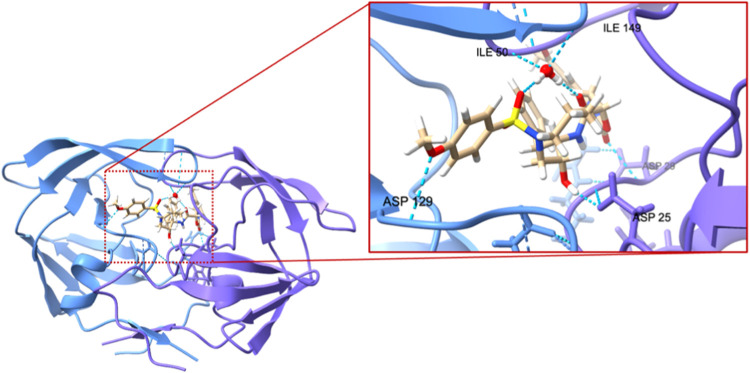
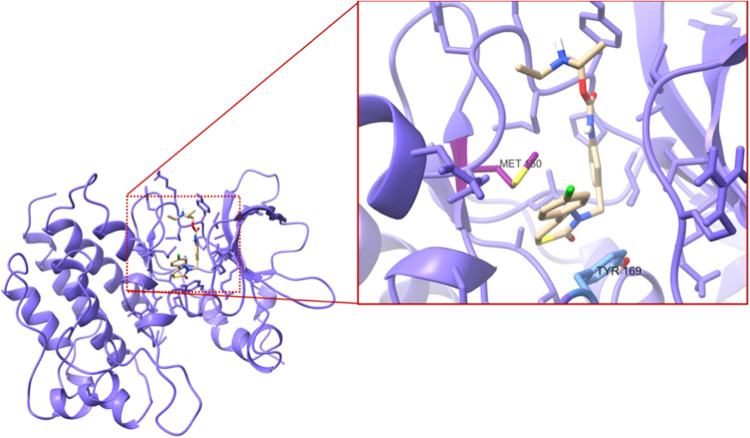
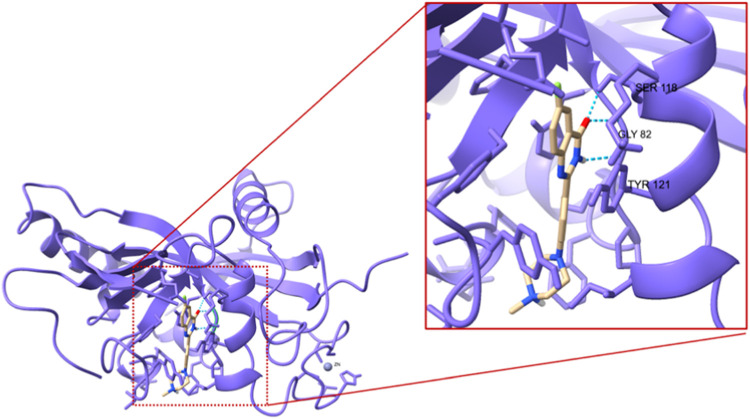
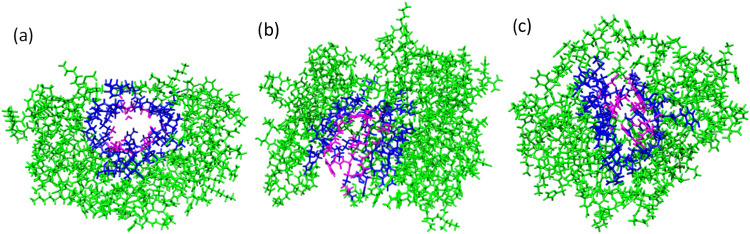
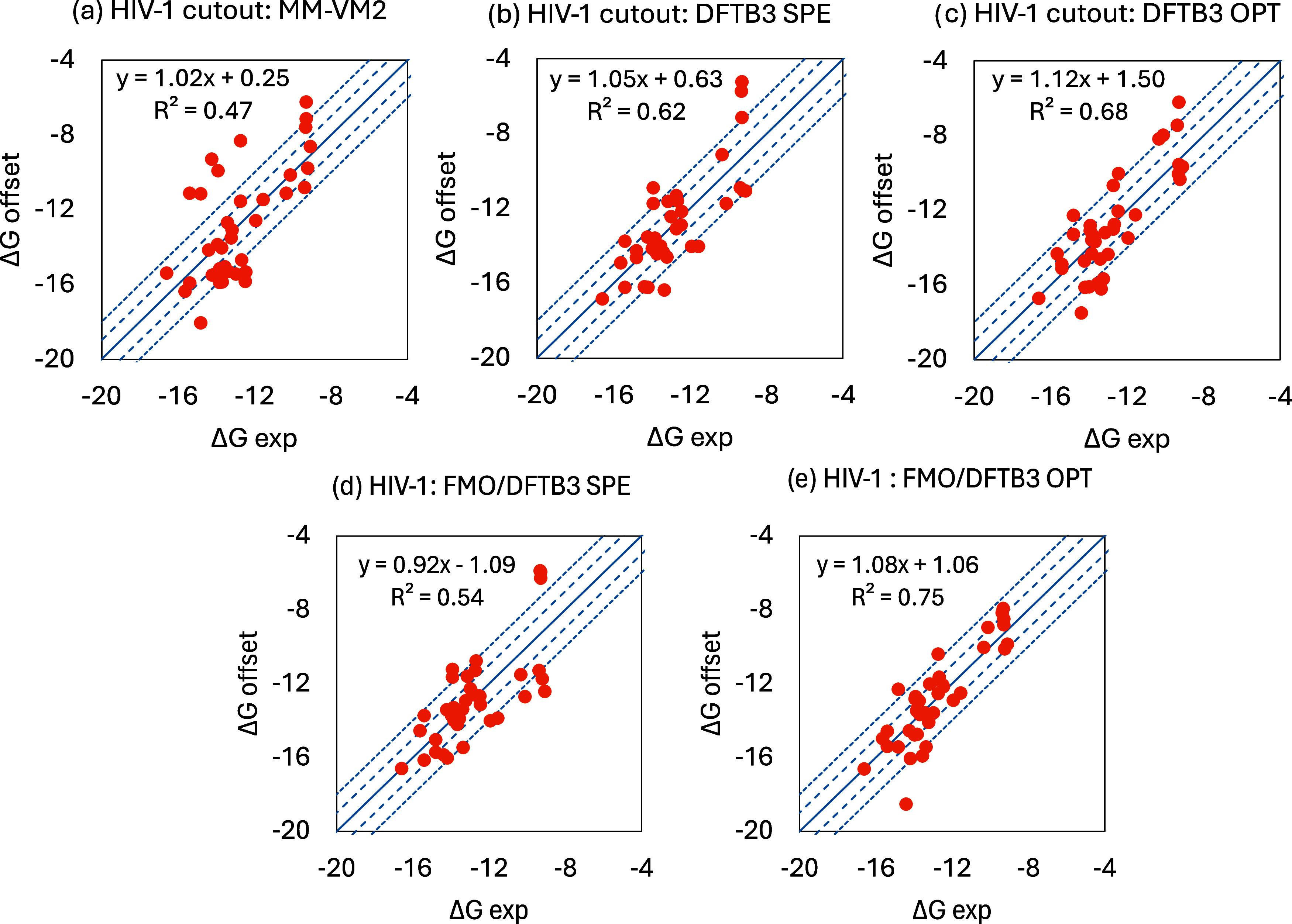
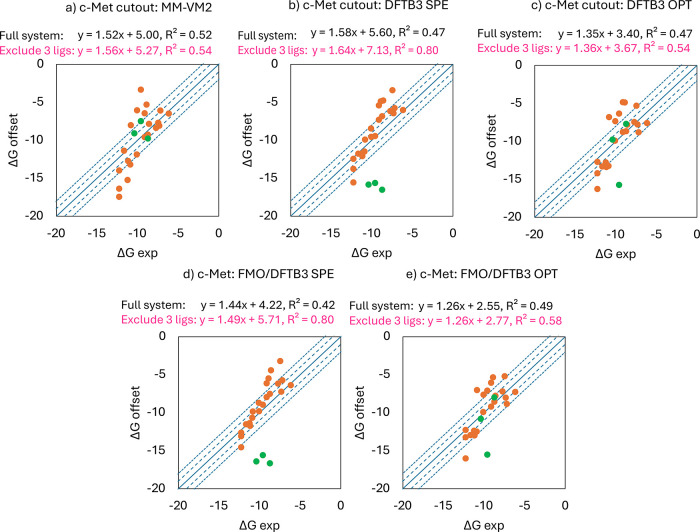
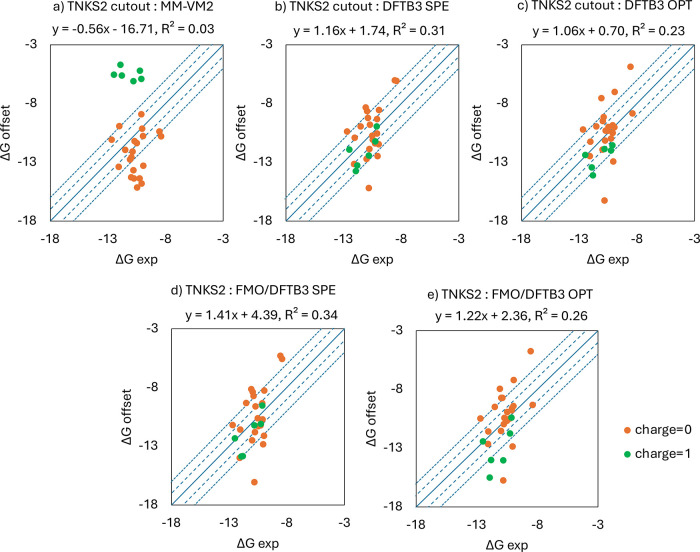
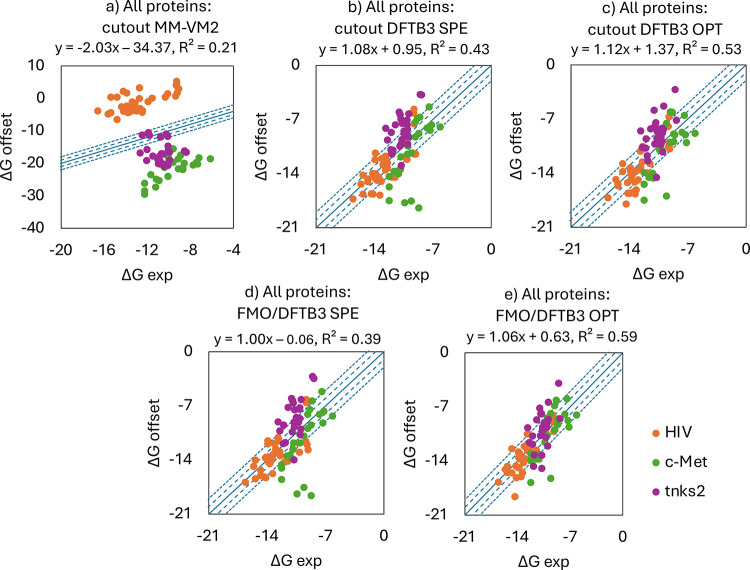
A new method, protein-ligand QM-VM2 (PLQM-VM2), to calculate protein-ligand binding free energies by combining mining minima, a statistical mechanics end-point-based approach, with quantum mechanical potentials is presented. PLQM-VM2 is described in terms of a highly flexible workflow that is initiated from a Protein Data Bank (PDB) file and a chemical structure data file (SD file) containing two-dimensional (2D) or three-dimensional (3D) ligand series coordinates. The workflow utilizes the previously developed molecular mechanics (MM) implementation of the second-generation mining minima method, MM-VM2, to provide ensembles of protein, free ligand, and protein-ligand conformers, which are postprocessed at chosen levels of QM theory, via the quantum chemistry software package GAMESS, to correct MM-based conformer geometries and electronic energies. The corrected energies are used in the calculation of configuration integrals, which on summation over the conformer ensembles give QM-corrected chemical potentials and ultimately QM-corrected binding free energies. In this work, PLQM-VM2 is applied to three benchmark protein-ligand series: HIV-1 protease/38 ligands, c-Met/24 ligands, and TNKS2/27 ligands. QM corrections are carried out at the semiempirical third-order density functional tight-binding level of theory, augmented with dispersion and damping corrections (DFTB3-D3(BJ)H). Bulk solvation effects are accounted for with the conductor-like polarizable continuum model (PCM). DFTB3-D3(BJ)H/PCM single-point energy-only and geometry optimization QM corrections are carried out in conjunction with two different models that address the large computational scaling associated with protein-sized molecular systems. One is a protein cutout model, whereby a set of protein atoms in and around the binding site are carved out, dangling bonds are capped with hydrogens, and only atoms directly in the protein binding site are mobile along with the ligand atoms. The other model is the Fragment Molecular Orbital (FMO) method, which includes the whole protein system but again only allows the binding site and ligand atoms to be mobile. All four of these methodological approaches to QM corrections provide significant improvement over MM-VM2 in terms of rank order and parametric linear correlation with experimentally determined binding affinities. Overall, FMO with geometry optimizations performed the best, but the much cheaper cutout single-point energy approach still provides a good level of accuracy. Furthermore, a clear result is that the PLQM-VM2 calculated binding free energies for the three diverse test systems in this work are, in contrast to those calculated using MM-VM2, directly comparable in energy scale. This suggests a basis for future development of a PLQM-VM2-based multiprotein screening capability to check for off-target activity of ligand series. Benchmark timings on a single compute node (32 CPU cores) for PLQM-VM2 calculation of the chemical potential of a protein-ligand complex range from ca. 30-45 min for the single-point energy approaches to ∼5 h for the cutout approach with geometry optimization and to ∼35 h for the full protein FMO approach with geometry optimization.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
-
- Schindler C. E. M.; Baumann H.; Blum A.; Böse D.; Buchstaller H.-P.; Burgdorf L.; Cappel D.; Chekler E.; Czodrowski P.; Dorsch D.; Eguida M. K. I.; Follows B.; Fuchß T.; Grädler U.; Gunera J.; Johnson T.; Jorand Lebrun C.; Karra S.; Klein M.; Knehans T.; Koetzner L.; Krier M.; Leiendecker M.; Leuthner B.; Li L.; Mochalkin I.; Musil D.; Neagu C.; Rippmann F.; Schiemann K.; Schulz R.; Steinbrecher T.; Tanzer E.-M.; Unzue Lopez A.; Viacava Follis A.; Wegener A.; Kuhn D. Large-Scale Assessment of Binding Free Energy Calculations in Active Drug Discovery Projects. J. Chem. Inf. Model. 2020, 60 (11), 5457–5474. 10.1021/acs.jcim.0c00900. - DOI - PubMed
-
- Reynolds C. H.Computer-Aided Drug Design: A Practical Guide to Protein-Structure-Based Modeling. In Drug Design: Structure- and Ligand-Based Approaches; Reynolds C. H.; Ringe D.; Merz J.; Kenneth M., Eds.; Cambridge University Press: Cambridge, 2010; pp 181–19610.1017/CBO9780511730412.014. - DOI
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous