

CPSF6 promotes HIV-1 preintegration complex function
- PMID: 40202316
- PMCID: PMC12090733
- DOI: 10.1128/jvi.00490-25
CPSF6 promotes HIV-1 preintegration complex function
Abstract
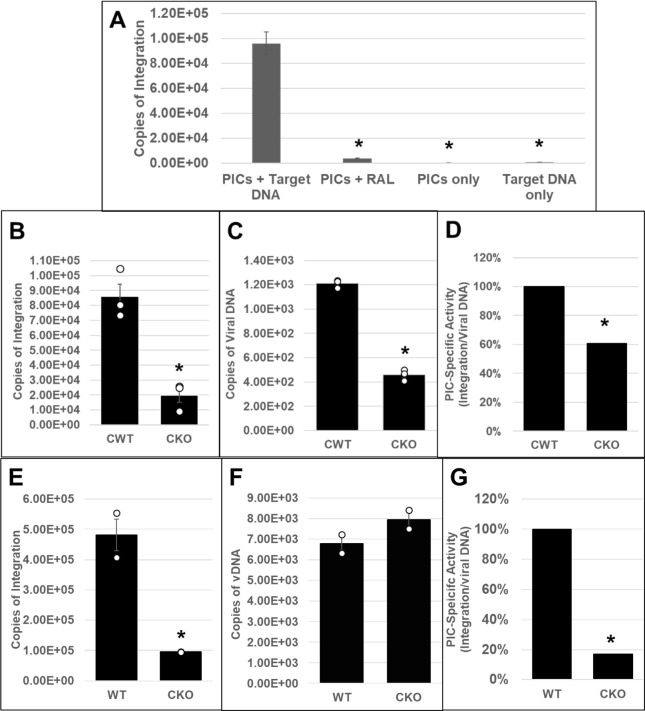
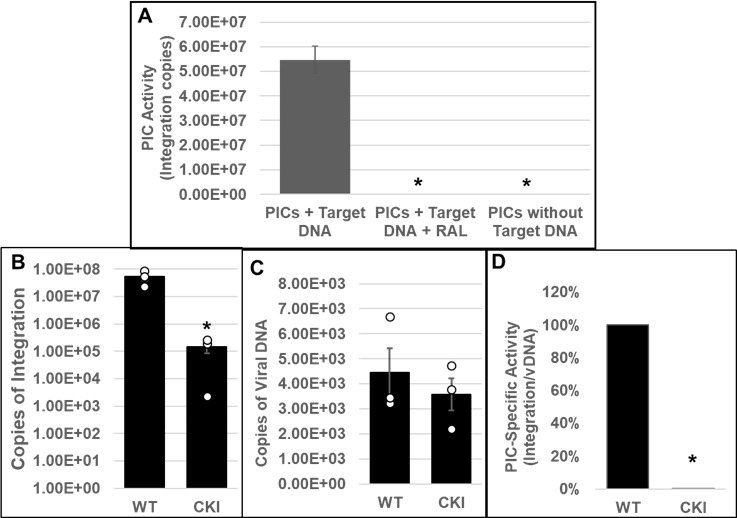
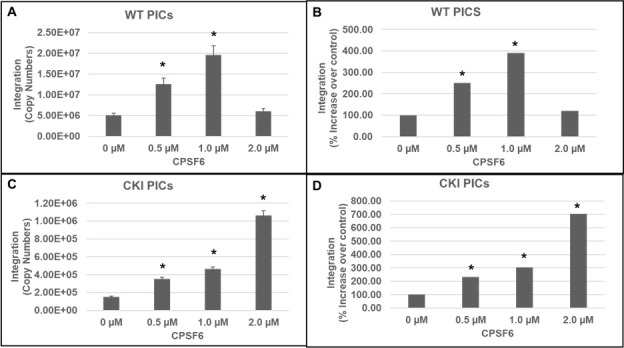
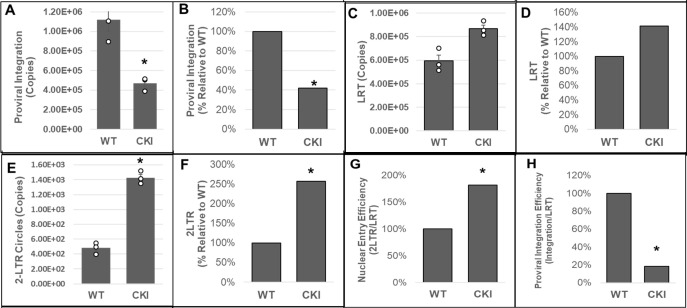
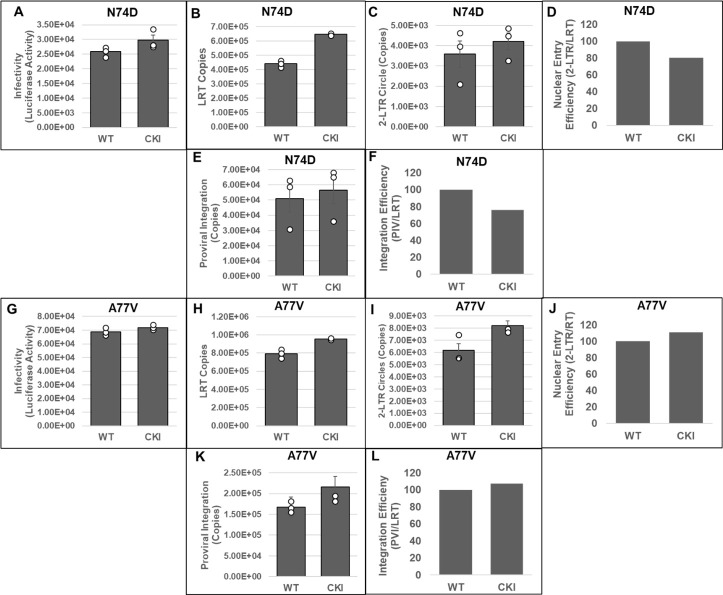
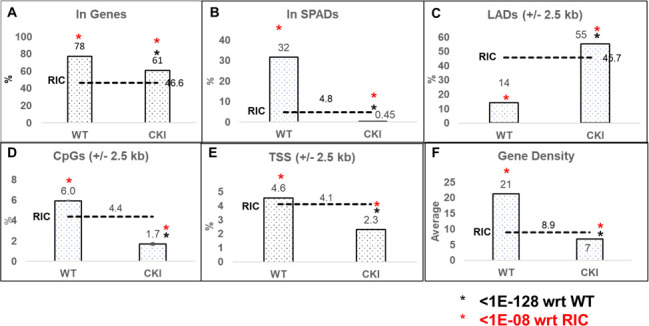
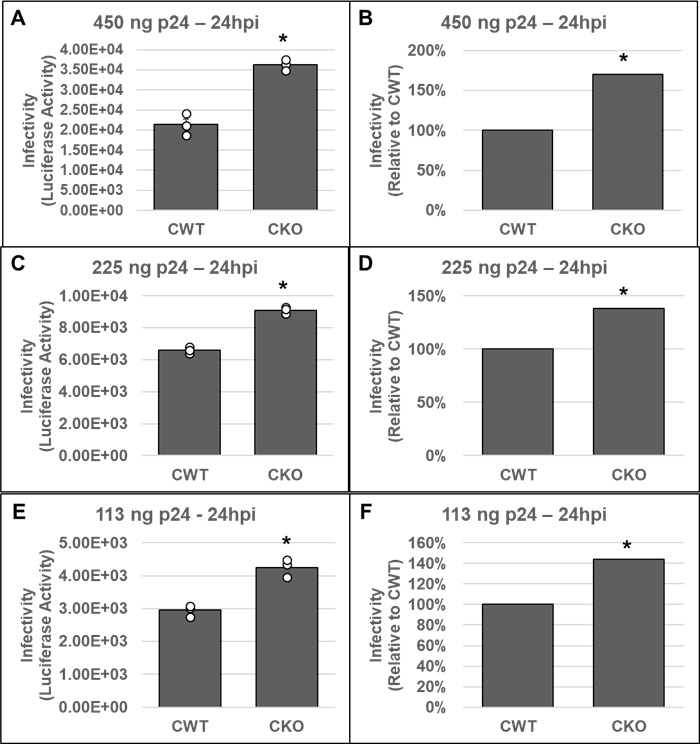
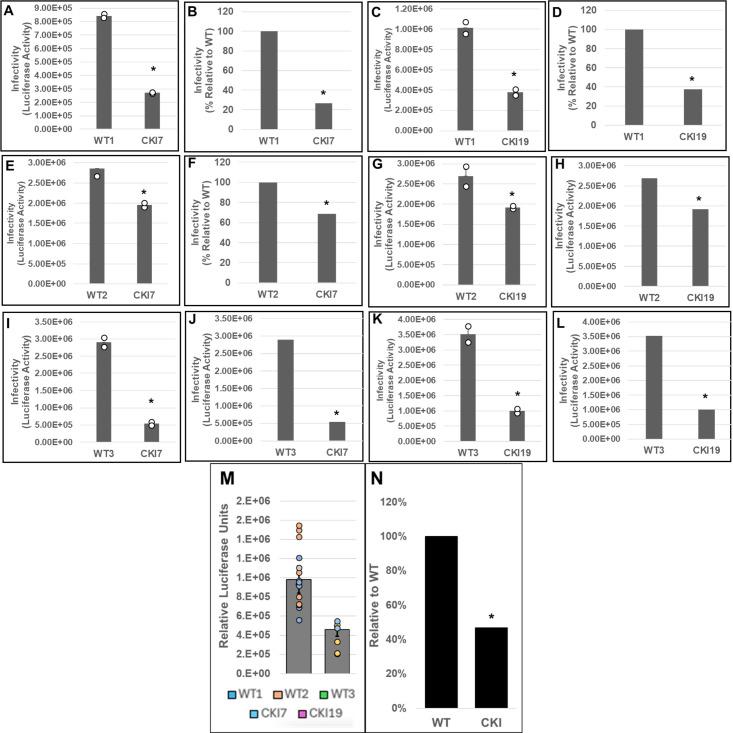
Cleavage and polyadenylation specificity factor 6 (CPSF6) is part of the cellular cleavage factor I mammalian (CFIm) complex that regulates mRNA processing and polyadenylation. CPSF6 also functions as an HIV-1 capsid (CA) binding host factor to promote viral DNA integration targeting into gene-dense regions of the host genome. However, the effects of CPSF6 on the activity of the HIV-1 preintegration complex (PIC)-the sub-viral machinery that carries out viral DNA integration-are unknown. To study CPSF6's role in HIV-1 PIC function, we extracted PICs from cells that are either depleted of CPSF6 or express a mutant form that cannot bind to CA. These PICs exhibited significantly lower viral DNA integration activity when compared to the control PICs. The addition of purified recombinant CPSF6 restored the integration activity of PICs extracted from the CPSF6-mutant cells, suggesting a direct role of CPSF6 in PIC function. To solidify CPSF6's role in PIC function, we inoculated CPSF6-depleted and CPSF6-mutant cells with HIV-1 particles and measured viral DNA integration into the host genome. A significant reduction in integration in these cells was detected, and this reduction was not a consequence of lower reverse transcription or nuclear entry. Additionally, mutant viruses deficient in CA-CPSF6 binding showed no integration defect in CPSF6-mutant cells. Finally, sequencing analysis revealed that HIV-1 integration into CPSF6-mutant cell genomes was significantly redirected away from gene-dense regions of chromatin compared to the control cells. Collectively, these results suggest that the CPSF6-CA interaction promotes PIC function both in vitro and in infected cells.IMPORTANCEHIV-1 infection is dependent on the interaction of the virus with cellular host factors. However, the molecular details of HIV-host factor interactions are not fully understood. For instance, the HIV-1 capsid provides binding interfaces for several host factors. CPSF6 is one such capsid-binding host factor, whose cellular function is to regulate mRNA processing and polyadenylation. Initial work identified a truncated cytosolic form of CPSF6 to restrict HIV infection by blocking viral nuclear entry. However, it is now established that the full-length CPSF6 primarily promotes HIV-1 integration targeting into gene-dense regions of the host genome. Here, we provide evidence that CPSF6-CA interaction stimulates the activity of HIV-1 preintegration complexes (PICs). We also describe that disruption of CPSF6-CA binding in target cells significantly reduces viral DNA integration and redirects integration targeting away from gene-dense regions into regions of low transcriptional activity. These findings identify a critical role for the CPSF6-CA interaction in PIC function and integration targeting.
Keywords: capsid; cleavage and polyadenylation specificity factor 6 (CPSF6); human immunodeficiency virus (HIV); integration; preintegration complex.
Conflict of interest statement
J.H. has received research support, paid to Northwestern University, from Gilead Sciences and is a paid consultant for Merck and Ridgeback Therapeutics.
Figures
References
-
- Ostrowski MA, Chun TW, Justement SJ, Motola I, Spinelli MA, Adelsberger J, Ehler LA, Mizell SB, Hallahan CW, Fauci AS. 1999. Both memory and CD45RA+/CD62L+ naive CD4(+) T cells are infected in human immunodeficiency virus type 1-infected individuals. J Virol 73:6430–6435. doi:10.1128/JVI.73.8.6430-6435.1999 - DOI - PMC - PubMed
-
- Brenchley JM, Hill BJ, Ambrozak DR, Price DA, Guenaga FJ, Casazza JP, Kuruppu J, Yazdani J, Migueles SA, Connors M, Roederer M, Douek DC, Koup RA. 2004. T-cell subsets that harbor human immunodeficiency virus (HIV) in vivo: implications for HIV pathogenesis. J Virol 78:1160–1168. doi:10.1128/jvi.78.3.1160-1168.2004 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
- R01 AI136740/AI/NIAID NIH HHS/United States
- R01 AI165236/AI/NIAID NIH HHS/United States
- T32 GM144927/GM/NIGMS NIH HHS/United States
- R56 AI122960/AI/NIAID NIH HHS/United States
- R01 DA057204/DA/NIDA NIH HHS/United States
- R01 AI052014/AI/NIAID NIH HHS/United States
- P30 AI110527/AI/NIAID NIH HHS/United States
- R25 AI164610/AI/NIAID NIH HHS/United States
- R37 AI039394/AI/NIAID NIH HHS/United States
- R01 AI162694/AI/NIAID NIH HHS/United States
- R01 AI170228/AI/NIAID NIH HHS/United States
- U54 MD007586/MD/NIMHD NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
