

Sawfish: improving long-read structural variant discovery and genotyping with local haplotype modeling
- PMID: 40203061
- PMCID: PMC12000528
- DOI: 10.1093/bioinformatics/btaf136
Sawfish: improving long-read structural variant discovery and genotyping with local haplotype modeling
Abstract
Motivation: Structural variants (SVs) play an important role in evolutionary and functional genomics but are challenging to characterize. High-accuracy, long-read sequencing can substantially improve SV characterization when coupled with effective calling methods. While state-of-the-art long-read SV callers are highly accurate, further improvements are achievable by systematically modeling local haplotypes during SV discovery and genotyping.
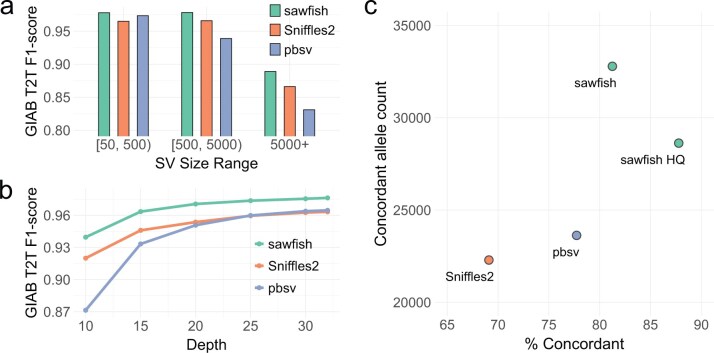
Results: We describe sawfish, an SV caller for mapped high-quality long reads incorporating systematic SV haplotype modeling to improve accuracy and resolution. Assessment against the draft Genome in a Bottle (GIAB) SV benchmark from the T2T-HG002-Q100 diploid assembly shows that sawfish has the highest accuracy among state-of-the-art long-read SV callers across every tested SV size group. Additionally, sawfish maintains the highest accuracy at every tested depth level from 10- to 32-fold coverage, such that other callers required at least 30-fold coverage to match sawfish accuracy at 15-fold coverage. Sawfish also shows the highest accuracy in the GIAB challenging medically relevant genes benchmark, demonstrating improvements in both comprehensive and medically relevant contexts.When joint-genotyping seven samples from CEPH-1463, sawfish has over 9000 more pedigree-concordant calls than other state-of-the-art SV callers, with the highest proportion of concordant SVs (81%). Sawfish's quality model enables selection for an even higher proportion of concordant SVs (88%), while still calling nearly 5000 more pedigree-concordant SVs than other callers. These results demonstrate that sawfish improves on the state-of-the-art for long-read SV calling accuracy across both individual and joint-sample analyses.
Availability and implementation: Sawfish source code, pre-compiled Linux binaries, and documentation are released on GitHub: https://github.com/PacificBiosciences/sawfish.
© The Author(s) 2025. Published by Oxford University Press.
Figures
Similar articles
-
NPSV-deep: a deep learning method for genotyping structural variants in short read genome sequencing data.Bioinformatics. 2024 Mar 4;40(3):btae129. doi: 10.1093/bioinformatics/btae129. Bioinformatics. 2024. PMID: 38444093 Free PMC article.
-
VolcanoSV enables accurate and robust structural variant calling in diploid genomes from single-molecule long read sequencing.Nat Commun. 2024 Aug 13;15(1):6956. doi: 10.1038/s41467-024-51282-0. Nat Commun. 2024. PMID: 39138168 Free PMC article.
-
SV-JIM, detailed pairwise structural variant calling using long-reads and genome assemblies.Methods. 2025 Feb;234:305-313. doi: 10.1016/j.ymeth.2024.12.015. Epub 2025 Jan 16. Methods. 2025. PMID: 39826659
-
A survey of algorithms for the detection of genomic structural variants from long-read sequencing data.Nat Methods. 2023 Aug;20(8):1143-1158. doi: 10.1038/s41592-023-01932-w. Epub 2023 Jun 29. Nat Methods. 2023. PMID: 37386186 Free PMC article. Review.
-
Structural variation detection using next-generation sequencing data: A comparative technical review.Methods. 2016 Jun 1;102:36-49. doi: 10.1016/j.ymeth.2016.01.020. Epub 2016 Feb 1. Methods. 2016. PMID: 26845461 Review.
Cited by
-
Human de novo mutation rates from a four-generation pedigree reference.Nature. 2025 Jul;643(8071):427-436. doi: 10.1038/s41586-025-08922-2. Epub 2025 Apr 23. Nature. 2025. PMID: 40269156 Free PMC article.
-
The Platinum Pedigree: a long-read benchmark for genetic variants.Nat Methods. 2025 Aug;22(8):1669-1676. doi: 10.1038/s41592-025-02750-y. Epub 2025 Aug 4. Nat Methods. 2025. PMID: 40759746
-
A Murine Database of Structural Variants Enables the Genetic Architecture of a Spontaneous Murine Lymphoma to be Characterized.bioRxiv [Preprint]. 2025 May 2:2025.01.09.632219. doi: 10.1101/2025.01.09.632219. bioRxiv. 2025. PMID: 39868308 Free PMC article. Preprint.
-
A Hitchhiker's Guide to long-read genomic analysis.Genome Res. 2025 Apr 14;35(4):545-558. doi: 10.1101/gr.279975.124. Genome Res. 2025. PMID: 40228901 Review.
References
-
- Kronenberg Z, Nolan C, Porubsky D et al. The platinum pedigree: a long-read benchmark for genetic variants. bioRxiv, 2024.10.02.616333, 2024, preprint: not peer reviewed.
MeSH terms
LinkOut - more resources
Full Text Sources
