

Perturb-tracing enables high-content screening of multi-scale 3D genome regulators
- PMID: 40211002
- PMCID: PMC12074983
- DOI: 10.1038/s41592-025-02652-z
Perturb-tracing enables high-content screening of multi-scale 3D genome regulators
Abstract
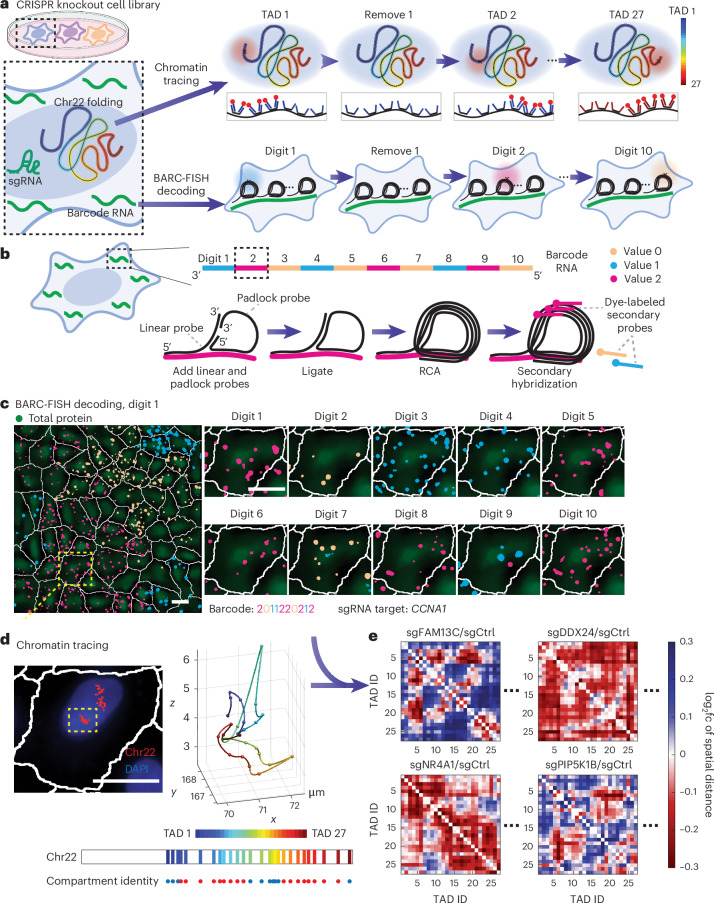
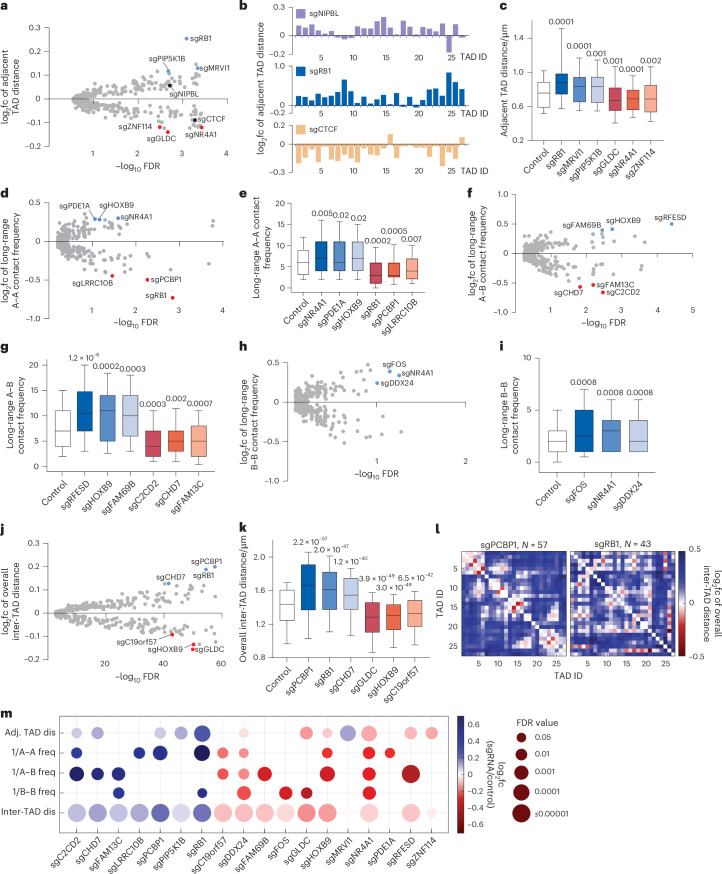
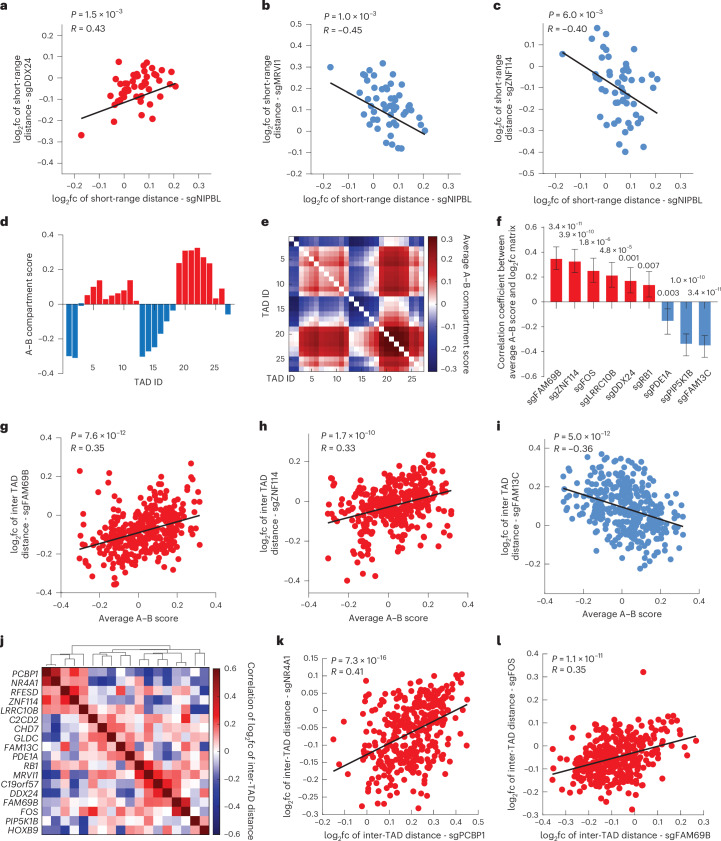
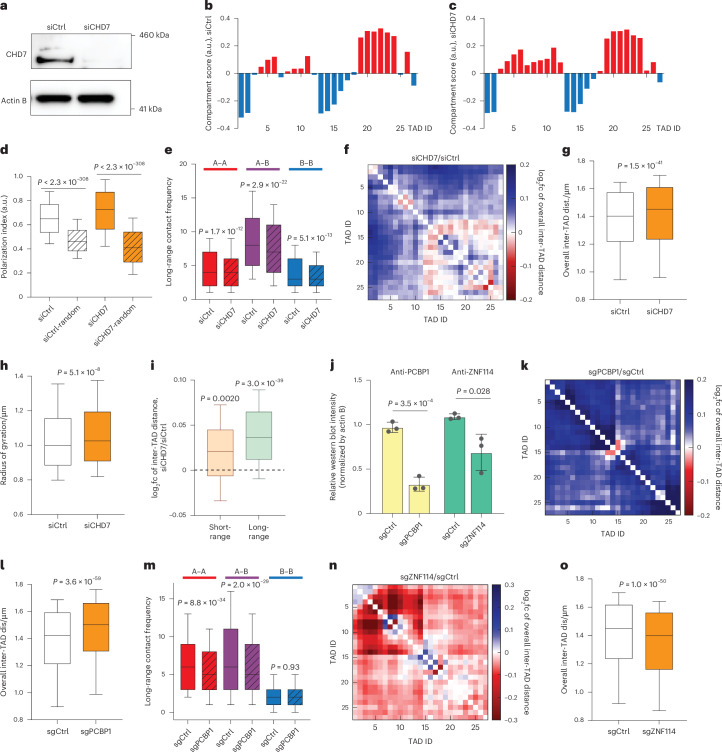
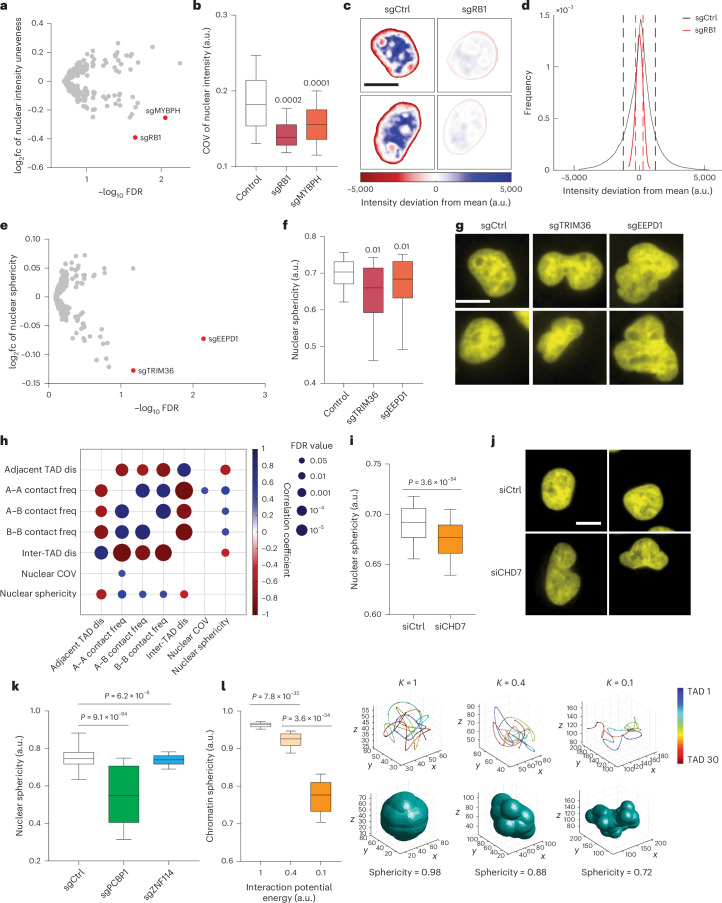
Three-dimensional (3D) genome organization becomes altered during development, aging and disease, but the factors regulating chromatin topology are incompletely understood and currently no technology can efficiently screen for new regulators of multi-scale chromatin organization. Here, we developed an image-based high-content screening platform (Perturb-tracing) that combines pooled CRISPR screens, a cellular barcode readout method (BARC-FISH) and chromatin tracing. We performed a loss-of-function screen in human cells, and visualized alterations to their 3D chromatin folding conformations, alongside perturbation-paired barcode readout in the same single cells. We discovered tens of new regulators of chromatin folding at different length scales, ranging from chromatin domains and compartments to chromosome territory. A subset of the regulators exhibited 3D genome effects associated with loop extrusion and A-B compartmentalization mechanisms, while others were largely unrelated to these known 3D genome mechanisms. Finally, we identified new regulators of nuclear architectures and found a functional link between chromatin compaction and nuclear shape. Altogether, our method enables scalable, high-content identification of chromatin and nuclear topology regulators that will stimulate new insights into the 3D genome.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: S.W., B.Y. and M.H. are inventors on a patent applied for by Yale University related to this work. The other authors declare no competing interests.
Figures







Update of
-
Perturb-tracing enables high-content screening of multiscale 3D genome regulators.bioRxiv [Preprint]. 2023 Nov 5:2023.01.31.525983. doi: 10.1101/2023.01.31.525983. bioRxiv. 2023. Update in: Nat Methods. 2025 May;22(5):950-961. doi: 10.1038/s41592-025-02652-z. PMID: 36778402 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
- UH3 CA268202/CA/NCI NIH HHS/United States
- R35 GM133712/GM/NIGMS NIH HHS/United States
- 2T32GM007499/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R01 HG012969/HG/NHGRI NIH HHS/United States
- R01 HG013503/HG/NHGRI NIH HHS/United States
- T32 GM007499/GM/NIGMS NIH HHS/United States
- 5T32GM007205/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- P50 CA196530/CA/NCI NIH HHS/United States
- U01 CA260701/CA/NCI NIH HHS/United States
- DP2GM137414/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R01 CA292936/CA/NCI NIH HHS/United States
- DP2 GM137414/GM/NIGMS NIH HHS/United States
- T32 GM007205/GM/NIGMS NIH HHS/United States
- R33 CA251037/CA/NCI NIH HHS/United States
- R01 HG011245/HG/NHGRI NIH HHS/United States
- R01HG013503/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
- R35GM133712/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
LinkOut - more resources
Full Text Sources
