

ATG16L1 restrains macrophage NLRP3 activation and alveolar epithelial cell injury during septic lung injury
- PMID: 40211890
- PMCID: PMC11986372
- DOI: 10.1002/ctm2.70289
ATG16L1 restrains macrophage NLRP3 activation and alveolar epithelial cell injury during septic lung injury
Abstract
Background: The lung is the organ most commonly affected by sepsis. Additionally, acute lung injury (ALI) resulting from sepsis is a major cause of death in intensive care units. Macrophages are essential for maintaining normal lung physiological functions and are implicated in various pulmonary diseases. An essential autophagy protein, autophagy-related protein 16-like 1 (ATG16L1), is crucial for the inflammatory activation of macrophages.
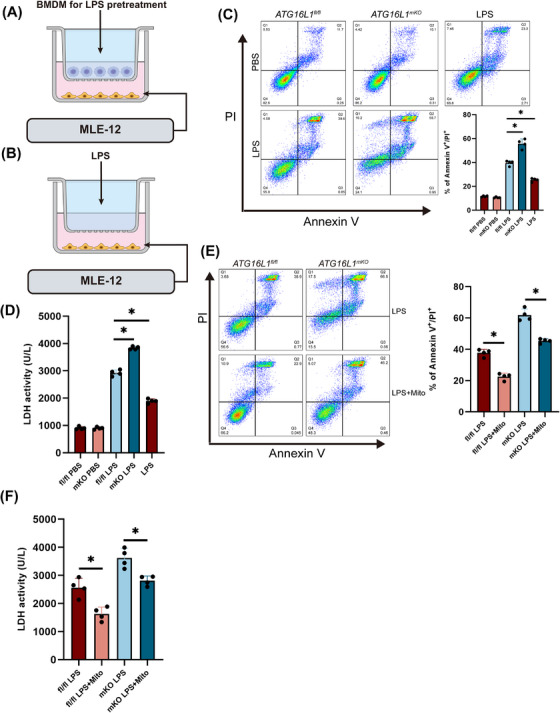
Methods: ATG16L1 expression was measured in lung from mice with sepsis. ALI was induced in myeloid ATG16L1-, NLRP3- and STING-deficient mice by intraperitoneal injection of lipopolysaccharide (LPS, 10 mg/kg). Using immunofluorescence and flow cytometry to assess the inflammatory status of LPS-treated bone marrow-derived macrophages (BMDMs). A co-culture system of BMDMs and MLE-12 cells was established in vitro.
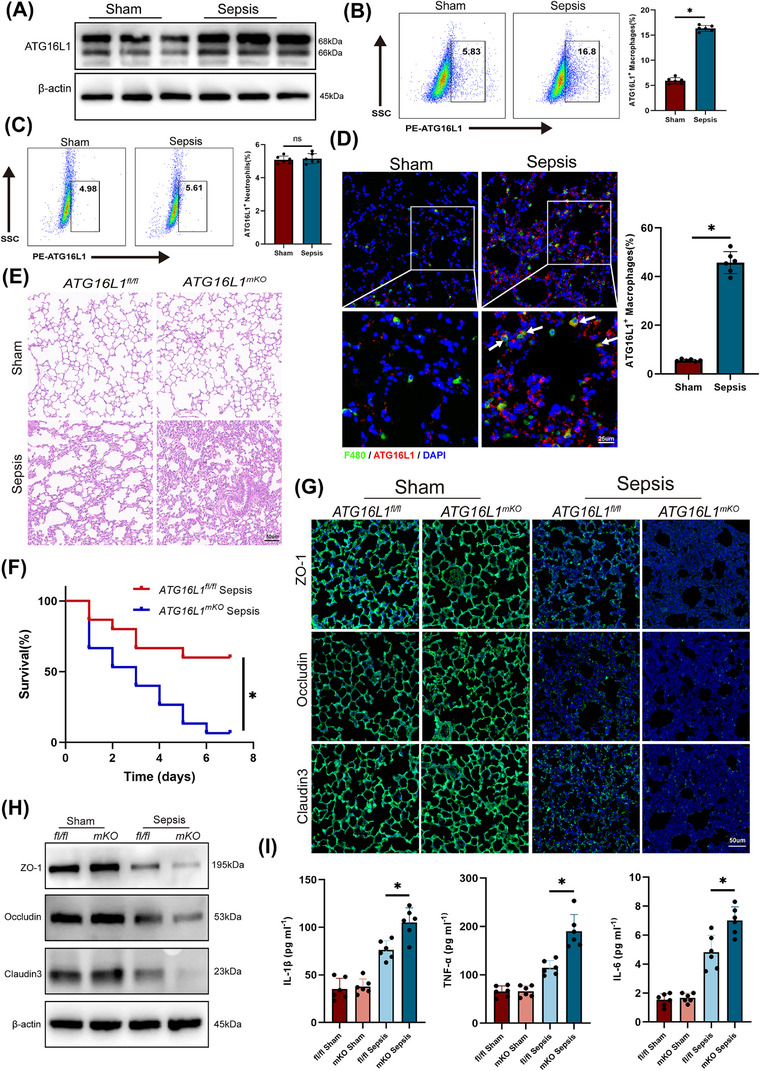
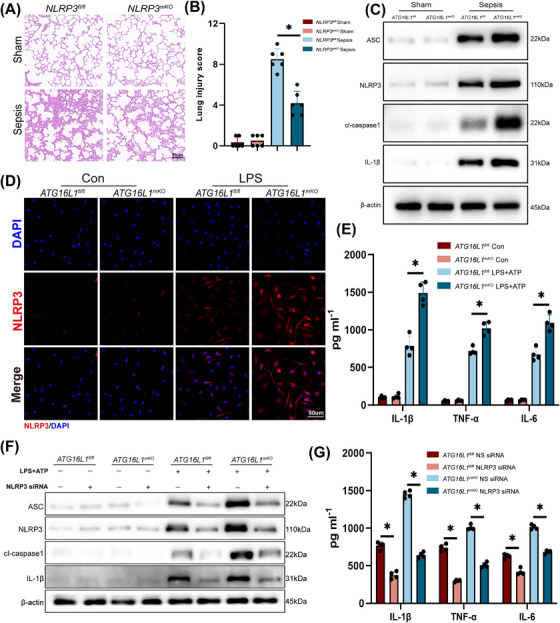
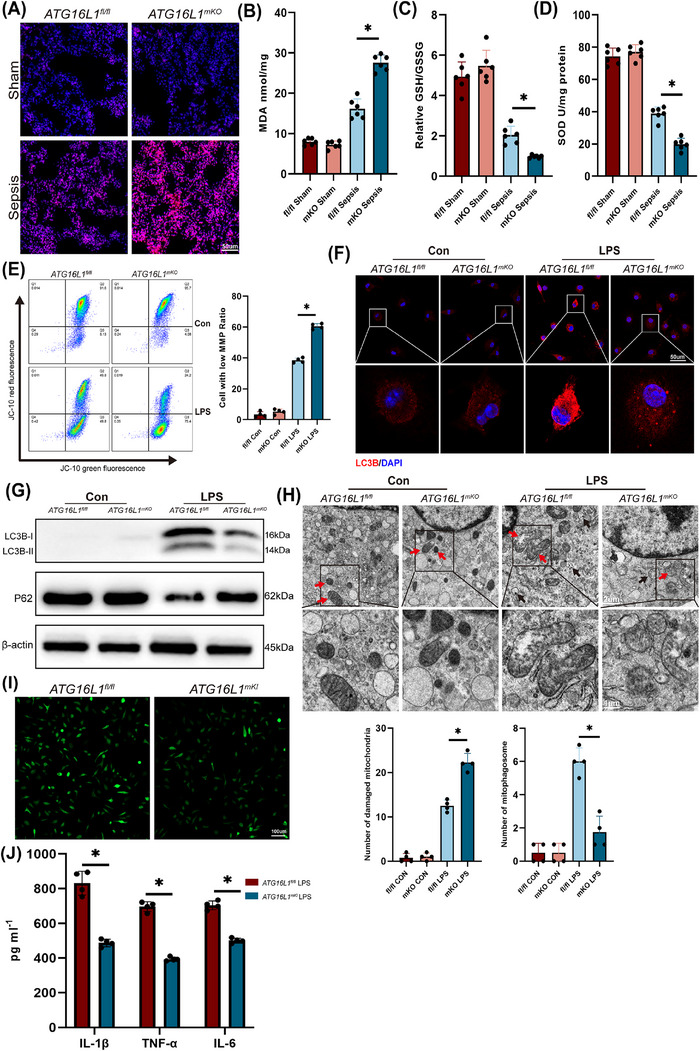
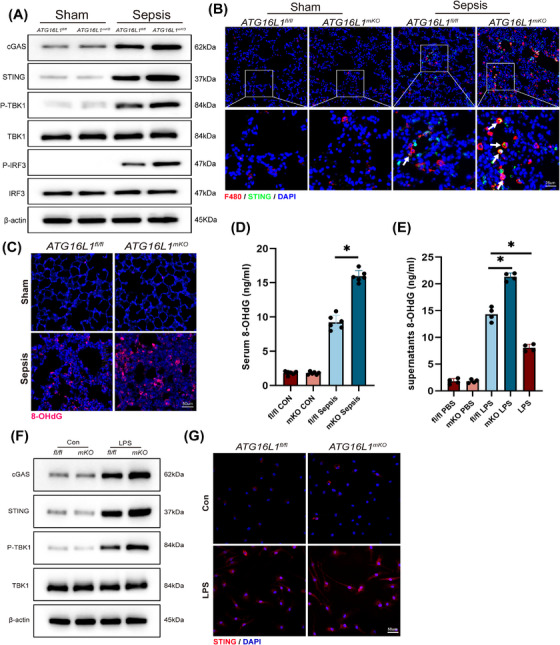
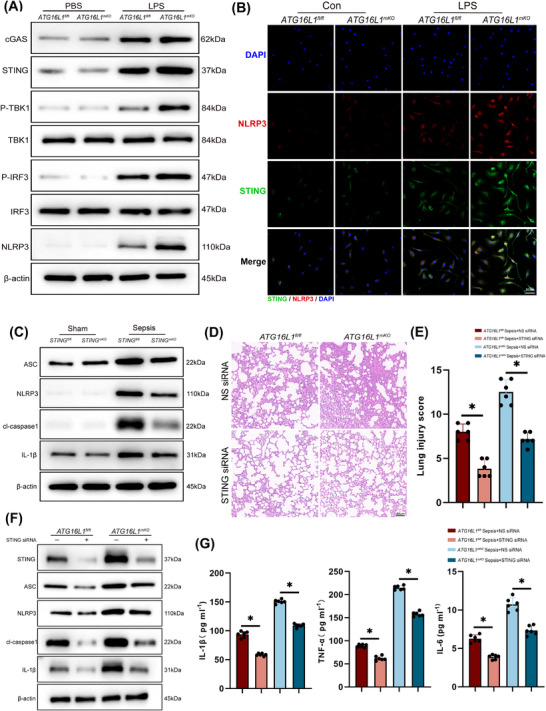
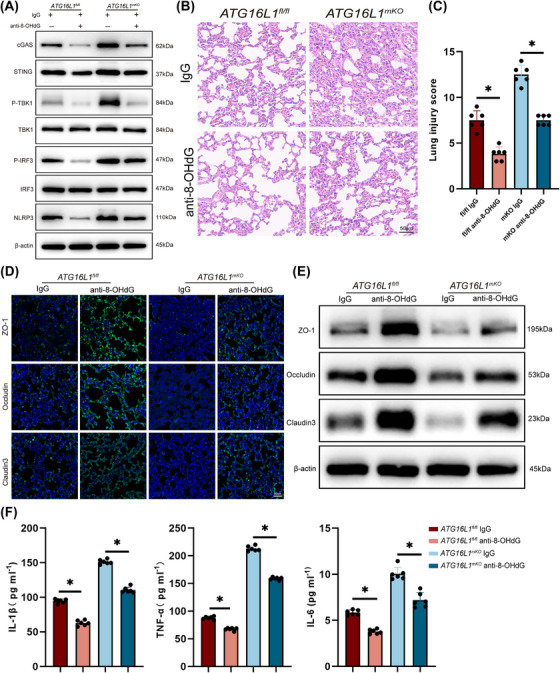
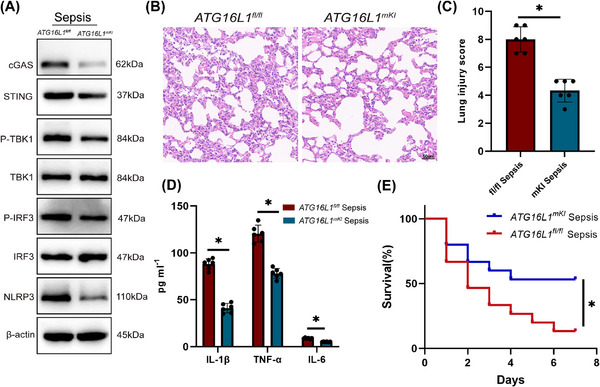
Results: Myeloid ATG16L1-deficient mice exhibited exacerbated septic lung injury and a more intense inflammatory response following LPS treatment. Mechanistically, ATG16L1-deficient macrophages exhibited impaired LC3B lipidation, damaged mitochondria and reactive oxygen species (ROS) accumulation. These abnormalities led to the activation of NOD-like receptor family pyrin domain-containing protein 3 (NLRP3), subsequently enhancing proinflammatory response. Overactivated ATG16L1-deficient macrophages aggravated the damage to alveolar epithelial cells and enhanced the release of double-stranded DNA (dsDNA), thereby promoting STING activation and subsequent NLRP3 activation in macrophages, leading to positive feedback activation of macrophage NLRP3 signalling. Scavenging mitochondrial ROS or inhibiting STING activation effectively suppresses NLRP3 activation in macrophages and alleviates ALI. Furthermore, overexpression of myeloid ATG16L1 limits NLRP3 activation and reduces the severity of ALI.
Conclusions: Our findings reveal a new role for ATG16L1 in regulating macrophage NLRP3 feedback activation during sepsis, suggesting it as a potential therapeutic target for treating sepsis-induced ALI.
Key points: Myeloid-specific ATG16L1 deficiency exacerbates sepsis-induced lung injury. ATG16L1-deficient macrophages exhibit impaired LC3B lipidation and ROS accumulation, leading to NLRP3 inflammasome activation. Uncontrolled inflammatory responses in ATG16L1-deficient macrophages aggravate alveolar epithelial cell damage. Alveolar epithelial cells release dsDNA, activating the cGAS-STING-NLRP3 signaling pathway, which subsequently triggers a positive feedback activation of NLRP3. Overexpression of ATG16L1 helps mitigate lung tissue inflammation, offering a novel therapeutic direction for sepsis-induced lung injury.
Keywords: ATG16L; NLRP3 inflammasome; STING; acute lung injury; macrophages.
© 2025 The Author(s). Clinical and Translational Medicine published by John Wiley & Sons Australia, Ltd on behalf of Shanghai Institute of Clinical Bioinformatics.
Conflict of interest statement
The authors declare they have no conflicts of interest.
Figures
Similar articles
-
NOD3 Reduces Sepsis-Induced Acute Lung Injury by Regulating the Activation of NLRP3 Inflammasome and the Polarization of Alveolar Macrophages.Inflammation. 2025 Aug;48(4):2395-2406. doi: 10.1007/s10753-024-02197-x. Epub 2024 Dec 2. Inflammation. 2025. PMID: 39621198
-
Mdivi-1 Attenuates Sepsis-Associated Acute Lung Injury by Inhibiting M1 Alveolar Macrophage Polarization and Pyroptosis.Mediators Inflamm. 2025 Mar 30;2025:3675276. doi: 10.1155/mi/3675276. eCollection 2025. Mediators Inflamm. 2025. PMID: 40196168 Free PMC article.
-
Gas6 induces AIM to suppress acute lung injury in mice by inhibiting NLRP3 inflammasome activation and inducing autophagy.Front Immunol. 2025 Feb 17;16:1523166. doi: 10.3389/fimmu.2025.1523166. eCollection 2025. Front Immunol. 2025. PMID: 40034700 Free PMC article.
-
SIRT1-Rab7 axis attenuates NLRP3 and STING activation through late endosomal-dependent mitophagy during sepsis-induced acute lung injury.Int J Surg. 2024 May 1;110(5):2649-2668. doi: 10.1097/JS9.0000000000001215. Int J Surg. 2024. PMID: 38445453 Free PMC article.
-
The role of lncRNAs in sepsis-induced acute lung injury: Molecular mechanisms and therapeutic potential.Arch Biochem Biophys. 2025 Jun;768:110407. doi: 10.1016/j.abb.2025.110407. Epub 2025 Apr 1. Arch Biochem Biophys. 2025. PMID: 40180295 Review.
Cited by
-
cGAS-STING targeting offers novel therapeutic regimen in sepsis-associated organ dysfunction.Cell Biol Toxicol. 2025 Jul 3;41(1):113. doi: 10.1007/s10565-025-10051-5. Cell Biol Toxicol. 2025. PMID: 40608126 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials