

Identification of novel variants in carbamoyl phosphate synthetase 1 gene and comparative pathogenicity assessments of CPS1 missense variants following ACMG/AMP-ClinGen recommendation for computational tools
- PMID: 40212732
- PMCID: PMC11982034
- DOI: 10.1016/j.ymgmr.2025.101208
Identification of novel variants in carbamoyl phosphate synthetase 1 gene and comparative pathogenicity assessments of CPS1 missense variants following ACMG/AMP-ClinGen recommendation for computational tools
Abstract
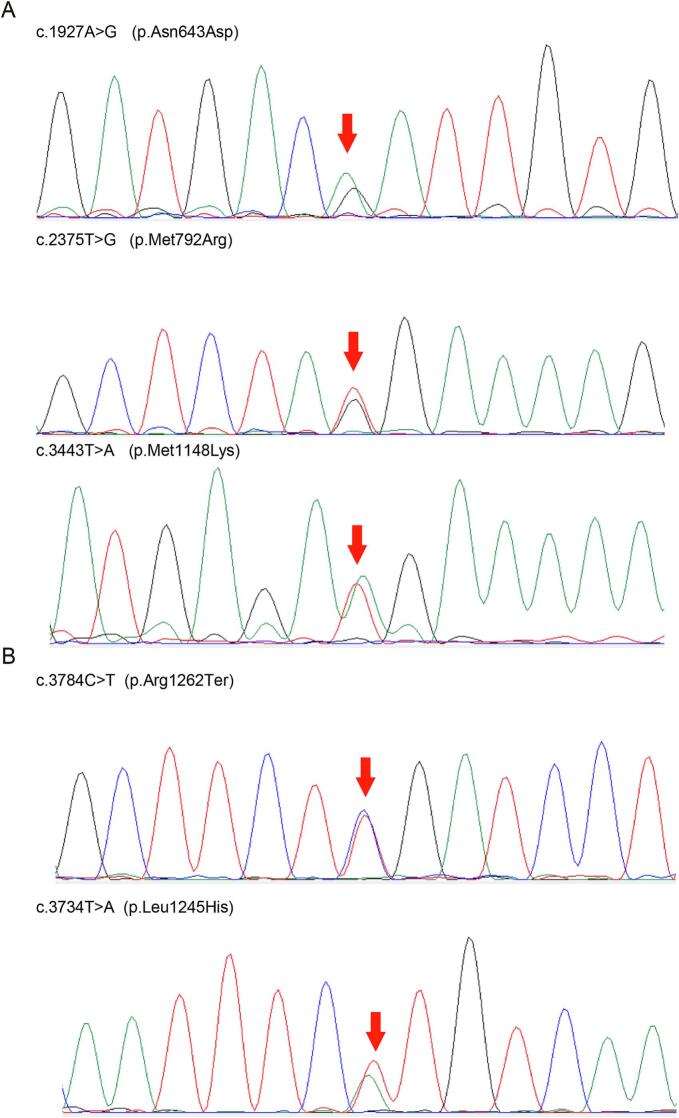
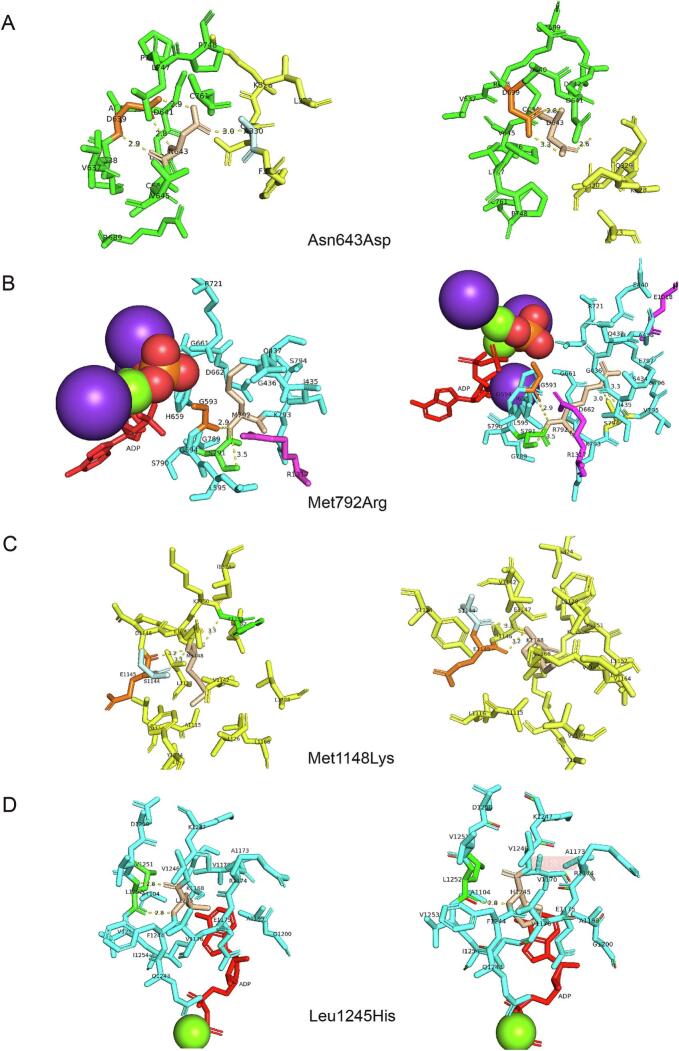
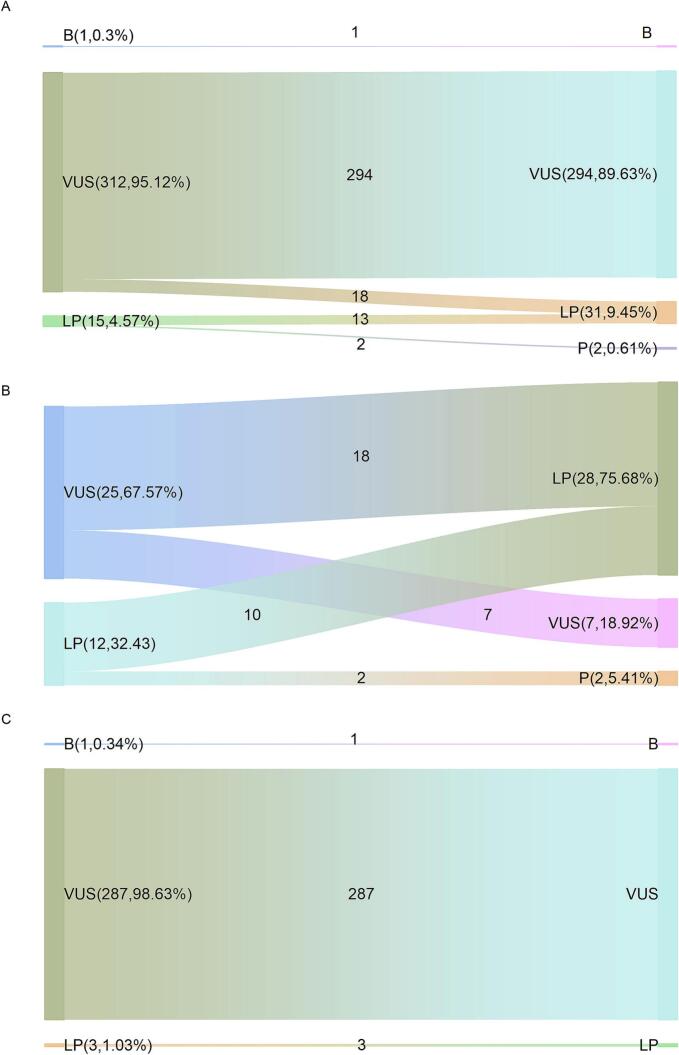
Carbamoyl phosphate synthetase I (CPS1) deficiency is a rare autosomal recessive metabolic abnormality cause by dysfunctionality of CPS1 and often result in unfavorable outcome. In this study, we presented the detailed laboratory features and genetic analysis of two patients with heterozygous variants of CPS1, c.1927 A > G (p.Asn643Asp), c.2375 T > G (p.Met792Arg), c.3443 T > A (p.Met1148Lys) in patient 1; c.3784C > T (p.Arg1262Ter), c.3734 T > A (p.Leu1245His) in patient 2, respectively. c.1927 A > G (p.Asn643Asp) and c.2375 T > G (p.Met792Arg) are novel out of 5 variants and classified as variants of uncertain significance (VUS) under the guidelines of ACMG/AMP-ClinGen. Structure-based analysis of 4 missense variants indicates deleterious alterations to the protein. Since the employment of genetic testing as a clinical diagnostic tool, distinguishing pathogenic from polymorphic changes poses significant problems for geneticists. As recommendation for PP3/BP4, the computational tools for missense variant have been published, we performed a comparative evaluation for pathogenicity interpretation in our patients and in ClinVar database regarding CPS1 missense variants under the updated guidelines of ACMG/AMP-ClinGen. The application of computational tools under the ACMG/AMP-ClinGen criteria revealed an increased sensitivity for pathogenicity evaluation, from variants of uncertain significance (VUS) to likely pathogenic (LP) in previously reported cases; while for variants without clinic information in the ClinVar database, the pathogenicity assessment of VUS remained, and shows a more optimistic and reliable clinical application in molecular diagnosis.
Keywords: Carbamoyl phosphate synthetase I deficiency; Missense variant; Pathogenicity interpretation; Urea cycle disorder.
© 2025 The Authors.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria.Am J Hum Genet. 2022 Dec 1;109(12):2163-2177. doi: 10.1016/j.ajhg.2022.10.013. Epub 2022 Nov 21. Am J Hum Genet. 2022. PMID: 36413997 Free PMC article.
-
ACMG/AMP interpretation of BRCA1 missense variants: Structure-informed scores add evidence strength granularity to the PP3/BP4 computational evidence.Am J Hum Genet. 2025 May 1;112(5):993-1002. doi: 10.1016/j.ajhg.2024.12.011. Epub 2025 Apr 14. Am J Hum Genet. 2025. PMID: 40233743 Free PMC article.
-
Carbamoyl phosphate synthetase 1 deficiency diagnosed by whole exome sequencing.J Clin Lab Anal. 2018 Feb;32(2):e22241. doi: 10.1002/jcla.22241. Epub 2017 Apr 26. J Clin Lab Anal. 2018. PMID: 28444906 Free PMC article.
-
Genetic, structural and biochemical basis of carbamoyl phosphate synthetase 1 deficiency.Mol Genet Metab. 2010 Dec;101(4):311-23. doi: 10.1016/j.ymgme.2010.08.002. Epub 2010 Aug 6. Mol Genet Metab. 2010. PMID: 20800523 Review.
-
Targeting CPS1 in the treatment of Carbamoyl phosphate synthetase 1 (CPS1) deficiency, a urea cycle disorder.Expert Opin Ther Targets. 2017 Apr;21(4):391-399. doi: 10.1080/14728222.2017.1294685. Epub 2017 Feb 20. Expert Opin Ther Targets. 2017. PMID: 28281899 Review.
References
-
- Keskinen P., Siitonen A., Salo M. Hereditary urea cycle diseases in Finland. Acta Paediatr. 2008;97(10):1412–1419. - PubMed
-
- Nagata N., Matsuda I., Oyanagi K. Estimated frequency of urea cycle enzymopathies in Japan. Am. J. Med. Genet. 1991;39(2):228–229. - PubMed
-
- Klaus V., Vermeulen T., Minassian B., et al. Highly variable clinical phenotype of carbamylphosphate synthetase 1 deficiency in one family: an effect of allelic variation in gene expression? Clin. Genet. 2009;76(3):263–269. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
