

Plasmodium falciparum infection induces T cell tolerance that is associated with decreased disease severity upon re-infection
- PMID: 40214640
- PMCID: PMC11987708
- DOI: 10.1084/jem.20241667
Plasmodium falciparum infection induces T cell tolerance that is associated with decreased disease severity upon re-infection
Abstract
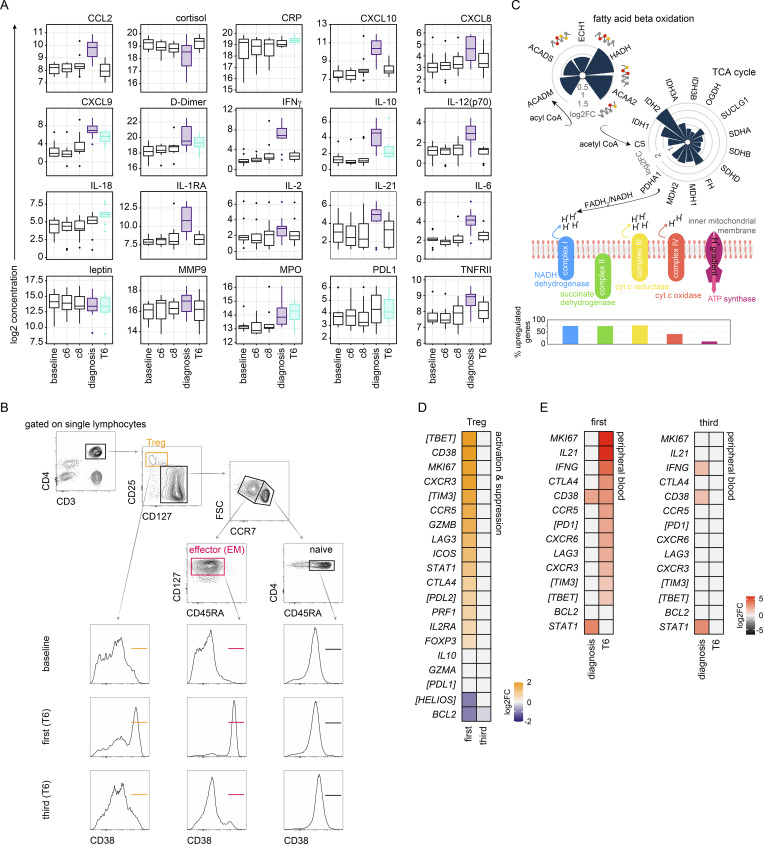
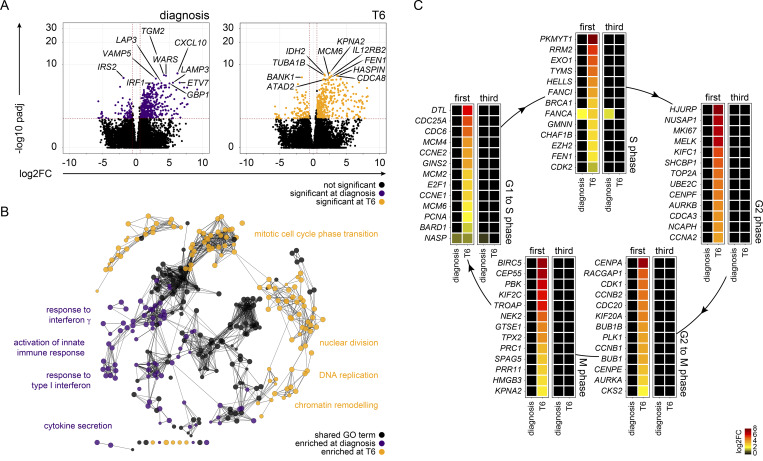
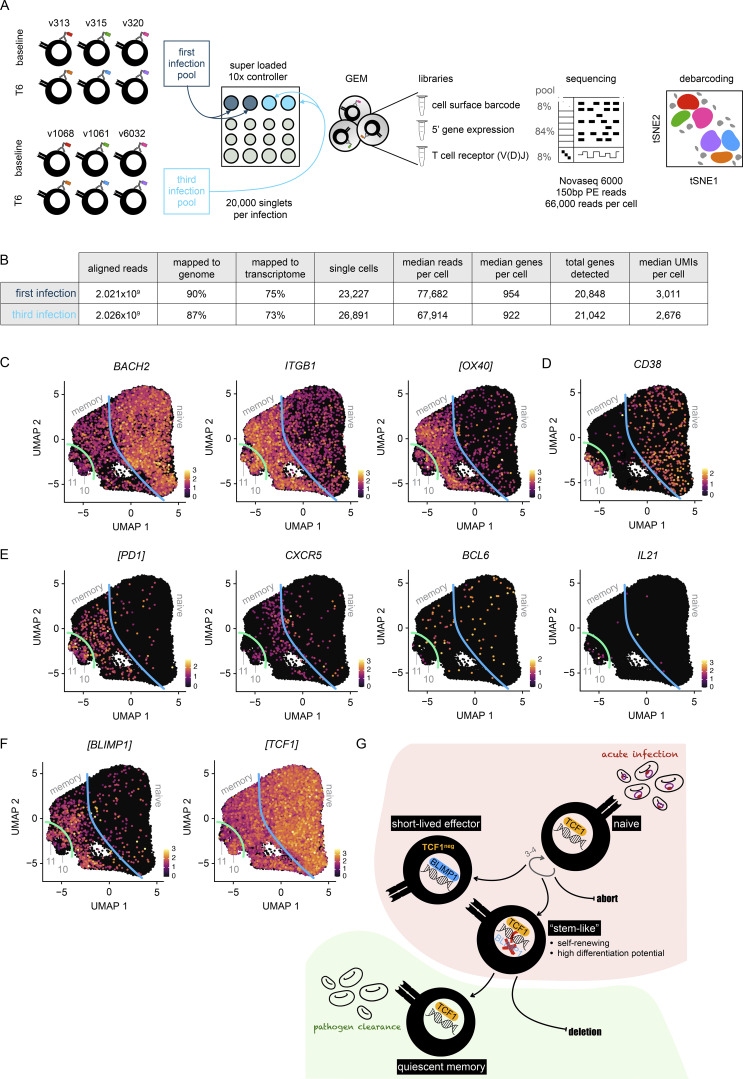
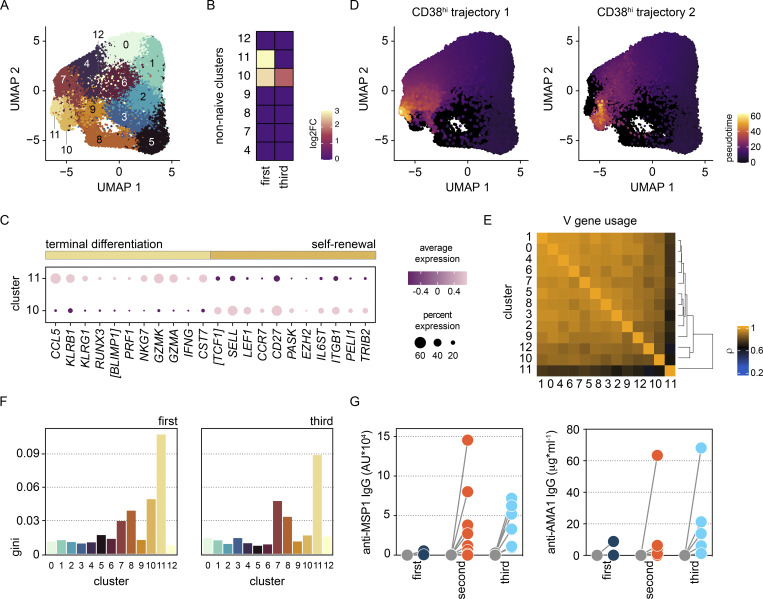
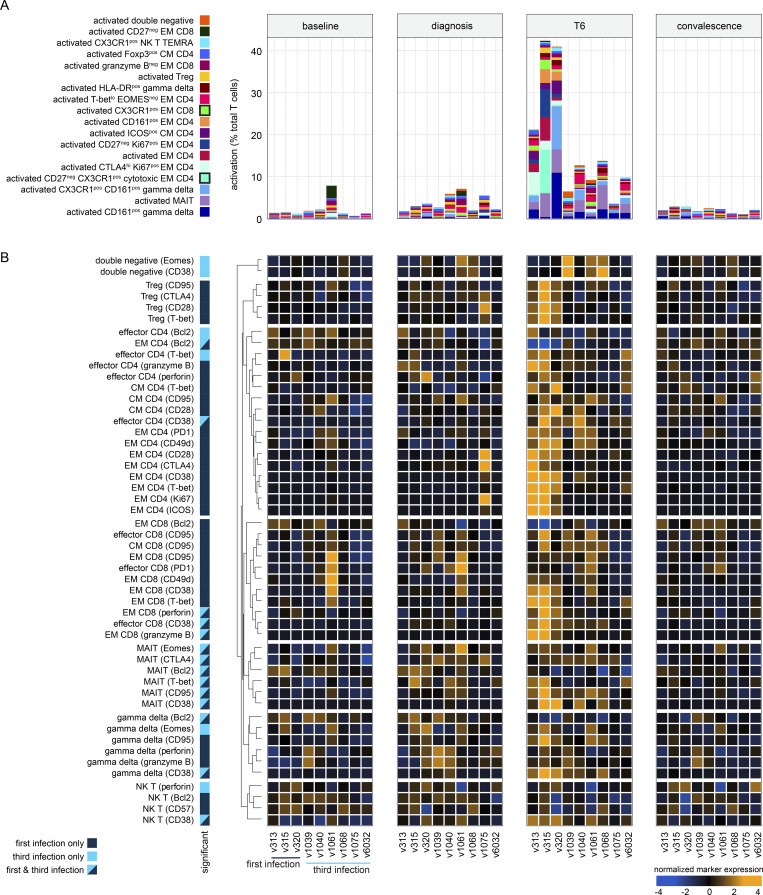
Immunity to severe malaria is acquired quickly, operates independently of pathogen load, and represents a highly effective form of disease tolerance. The mechanism that underpins tolerance remains unknown. We used a human rechallenge model of falciparum malaria in which healthy adult volunteers were infected three times over a 12 mo period to track the development of disease tolerance in real-time. We found that parasitemia triggered a hardwired innate immune response that led to systemic inflammation, pyrexia, and hallmark symptoms of clinical malaria across the first three infections of life. In contrast, a single infection was sufficient to reprogram T cell activation and reduce the number and diversity of effector cells upon rechallenge. Crucially, this did not silence stem-like memory cells but instead prevented the generation of cytotoxic effectors associated with autoinflammatory disease. Tolerized hosts were thus able to prevent collateral tissue damage in the absence of antiparasite immunity.
© 2025 Muñoz Sandoval et al.
Conflict of interest statement
Disclosures: The authors declare no competing interests exist.
Figures







References
-
- Antonelli, L.R., Leoratti F.M., Costa P.A., Rocha B.C., Diniz S.Q., Tada M.S., Pereira D.B., Teixeira-Carvalho A., Golenbock D.T., Gonçalves R., and Gazzinelli R.T.. 2014. The CD14+CD16+ inflammatory monocyte subset displays increased mitochondrial activity and effector function during acute Plasmodium vivax malaria. PLoS Pathog. 10:e1004393. 10.1371/journal.ppat.1004393 - DOI - PMC - PubMed
MeSH terms
Grants and funding
- 733073/European Union's Horizon 2020
- 203764/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- European Union
- Human Infection Challenge Network for Vaccine Development
- BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- University of Edinburgh
- AID-OAA-C-15-00071/U.S. Agency for International Development
- Jenner Institute
- 107668/Z/15/Z/Royal Society
- National Institute for Health Research Oxford Biomedical Research Centre
- WT_/Wellcome Trust/United Kingdom
- Darwin Trust of Edinburgh
- MRC_/Medical Research Council/United Kingdom
- Global Challenges Research Fund
LinkOut - more resources
Full Text Sources
