

Novel variants of SYNGAP1 associated epileptic encephalopathy: two cases report and literature review
- PMID: 40217345
- PMCID: PMC11960226
- DOI: 10.1186/s42494-022-00114-z
Novel variants of SYNGAP1 associated epileptic encephalopathy: two cases report and literature review
Abstract
Background: SYNGAP1 is a significant genetic risk factor for global developmental delay, autism spectrum disorder, and epileptic encephalopathy. De novo loss-of-function variants in this gene cause a neurodevelopmental disorder, for example, early-onset and drug-refractory seizures. We report two children with global developmental delay and epileptic encephalopathy, which are caused by SYNGAP1 gene novel mutations, and drug treatment is effective.
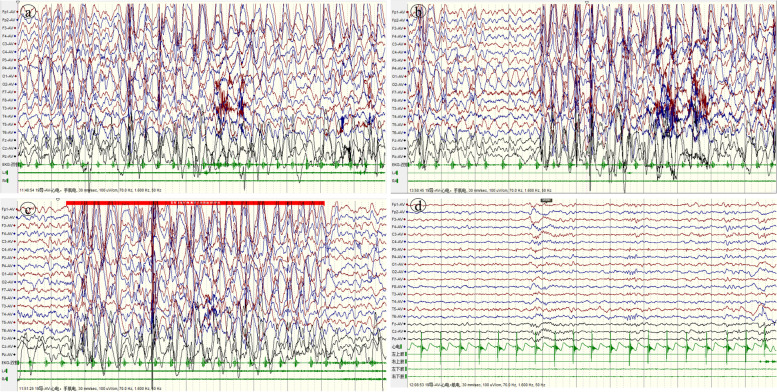
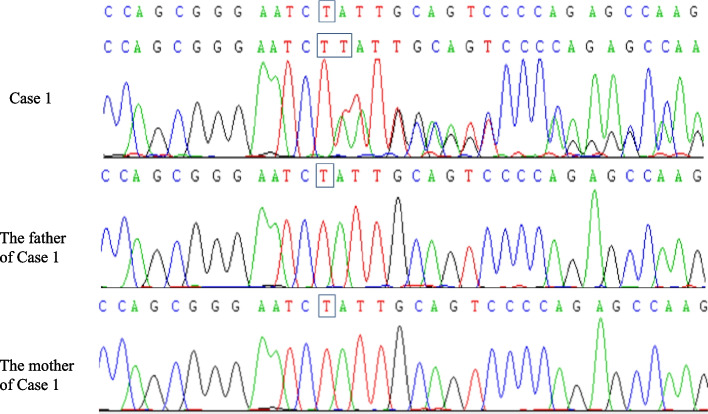
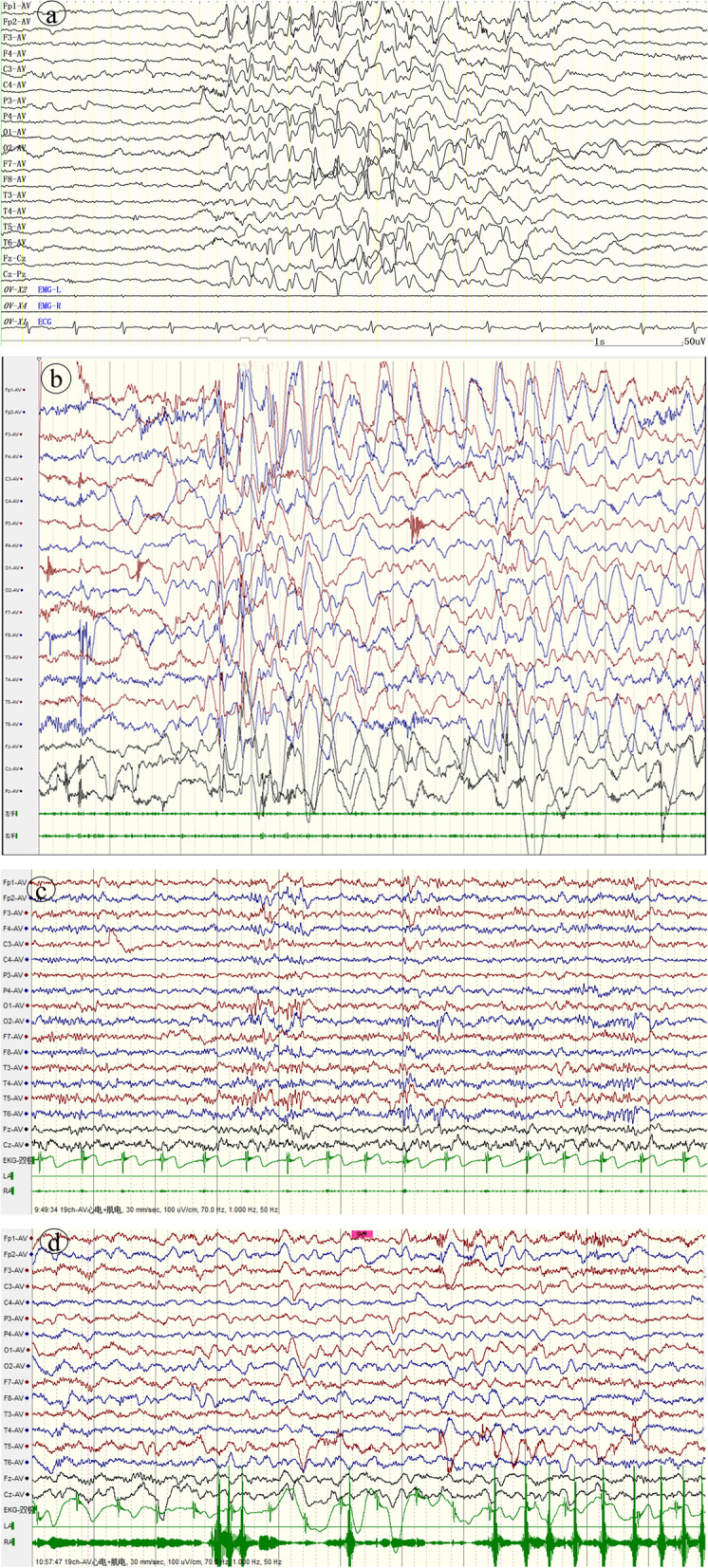
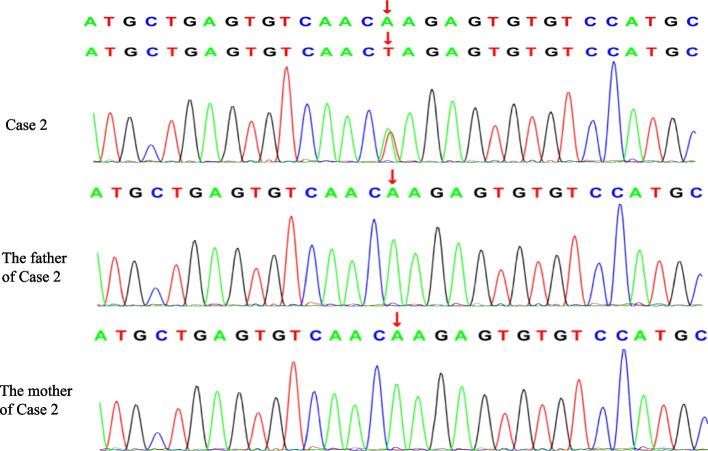
Case presentation: We report a boy and a girl presented with global developmental delay when they were young babies; as they grew up, cognitive impairment and social-communication disorder became more and more prominent; unfortunately, the patients developed into various seizure types, including eyelid myoclonia, myoclonic and absences when the boy was 1 year 8 mouths old and the girl was 3 years old. The two patients were found two previously unknown mutations by high throughput sequencing [c.3271_ c.3272insT; (p.L1091L fs*62), c.2515A > T (p.K839*)] in exon 15 of the SYNGAP in the proband. Sanger sequencing confirmed the heterozygous nature, and neither of their parents carried the same mutation. The girl treated with valproic acid and prednisone became seizure-free, and valproic acid and levetiracetam combined with clonazepam were influential in the other.
Conclusions: The global developmental delay and epileptic encephalopathy of the children were probably due to the pathogenic mutation of the SYNGAP1 gene, and prednisone and clonazepam may be effective in achieving seizure-free.
Keywords: SYNGAP1; Case report; Clonazepam; Epilepsy; Prednisone.
© 2023. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Written informed consent was obtained from the children’s guardians to publish their cases, and the Research Ethics Committee of Children's Hospital of Jiangxi Province provided approval for this study (approval number is JXSETYY-YXKY-20210035). Consent for publication: Written informed consent was obtained from the guardians of the participants for the publication of this case report. Competing interests: None of the authors declared any conflict of interest.
Figures

Similar articles
-
SYNGAP1 encephalopathy: A distinctive generalized developmental and epileptic encephalopathy.Neurology. 2019 Jan 8;92(2):e96-e107. doi: 10.1212/WNL.0000000000006729. Epub 2018 Dec 12. Neurology. 2019. PMID: 30541864 Free PMC article.
-
Genetic and neurodevelopmental spectrum of SYNGAP1-associated intellectual disability and epilepsy.J Med Genet. 2016 Aug;53(8):511-22. doi: 10.1136/jmedgenet-2015-103451. Epub 2016 Mar 17. J Med Genet. 2016. PMID: 26989088
-
Add-on cannabidiol significantly decreases seizures in 3 patients with SYNGAP1 developmental and epileptic encephalopathy.Epilepsia Open. 2020 Jul 1;5(3):496-500. doi: 10.1002/epi4.12411. eCollection 2020 Sep. Epilepsia Open. 2020. PMID: 32913957 Free PMC article.
-
[Early infantile epileptic encephalopathy caused by PACS2 gene variation: three cases report and literature review].Zhonghua Er Ke Za Zhi. 2021 Jul 2;59(7):594-599. doi: 10.3760/cma.j.cn112140-20201122-01047. Zhonghua Er Ke Za Zhi. 2021. PMID: 34405643 Review. Chinese.
-
Roadmap to advance therapeutics for SYNGAP1-related disorder: a patient organization perspective from SynGAP Research Fund.Ther Adv Rare Dis. 2025 Jan 12;6:26330040241308285. doi: 10.1177/26330040241308285. eCollection 2025 Jan-Dec. Ther Adv Rare Dis. 2025. PMID: 39807402 Free PMC article. Review.
References
-
- Mignot C, Stülpnagel Cv, Nava C, Ville D, Sanlaville D, Lesca G, et al. Genetic and neurodevelopmental spectrum of SYNGAP1-associated intellectual disability and epilepsy. J Med Genet. 2016;53(8):511–22. - PubMed
LinkOut - more resources
Full Text Sources