

Biomolecular mechanisms of epileptic seizures and epilepsy: a review
- PMID: 40217521
- PMCID: PMC11960269
- DOI: 10.1186/s42494-023-00137-0
Biomolecular mechanisms of epileptic seizures and epilepsy: a review
Abstract
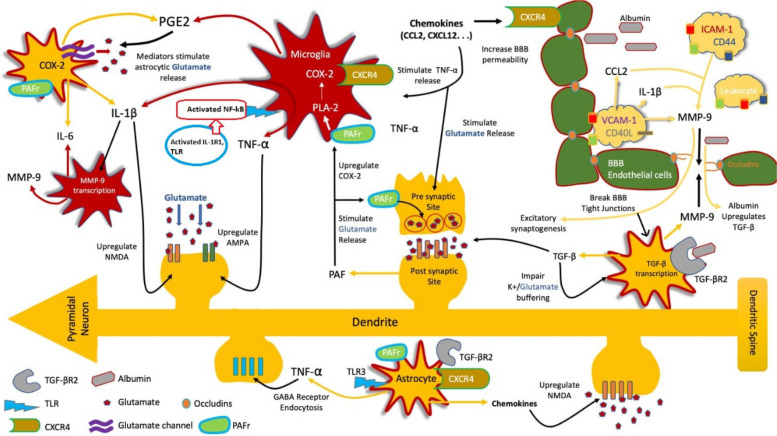
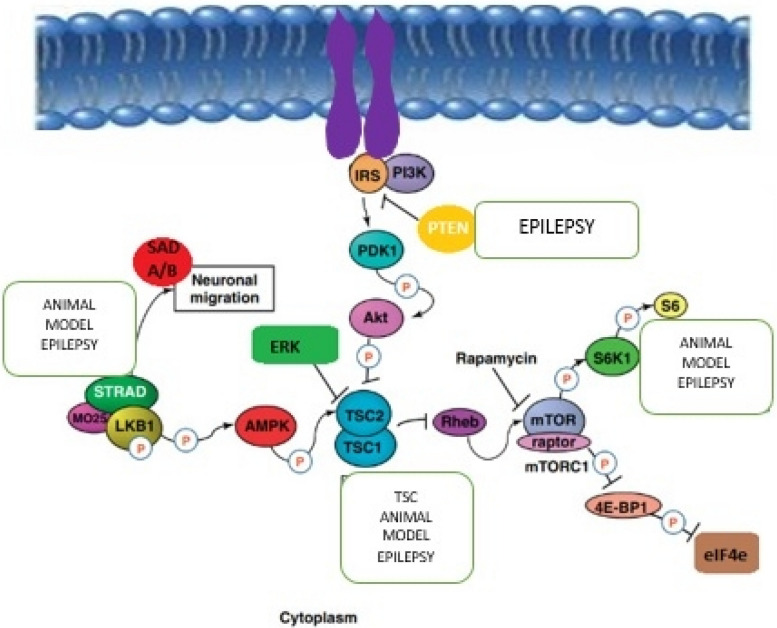
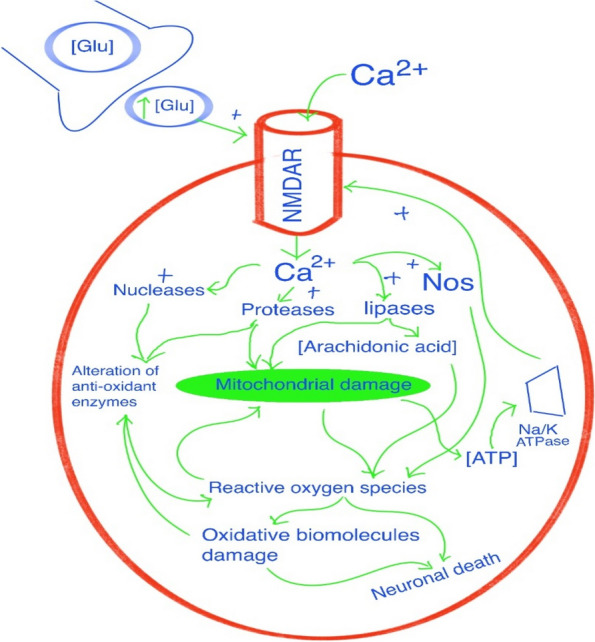
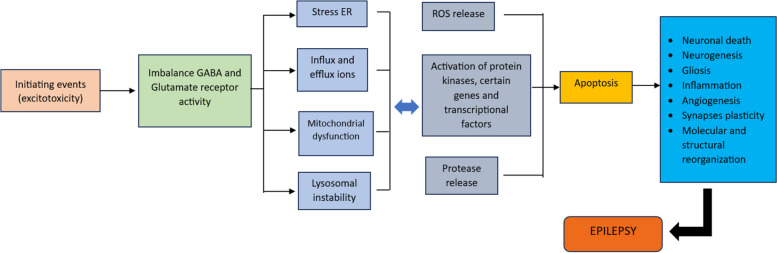
Epilepsy is a recurring neurological disease caused by the abnormal electrical activity in the brain. This disease has caused about 50 new cases in 100,000 populations every year with the clinical manifestations of awareness loss, bruising, and mobility abnormalities. Due to the lack understanding of the pathophysiology behind the illness, a wide variety of medications are available to treat epilepsy. Epileptogenesis is the process by which a normally functioning brain undergoes alterations leading to the development of epilepsy, involving various factors. This is related to the inflammation which is driven by cytokines like IL-1 and tumor necrosis factor-α (TNF-α) leads to neuronal hyperexcitability. Pro-inflammatory cytokines from activated microglia and astrocytes in epileptic tissue initiate an inflammatory cascade, heightening neuronal excitability and triggering epileptiform activity. The blood-brain barrier (BBB) maintains central nervous system integrity through its tight endothelial connections, but inflammation impact BBB structure and function which leads to immune cell infiltration. The mammalian target of rapamycin (mTOR) pathway's excessive activation influences epileptogenesis, impacting neuronal excitability, and synapse formation, with genetic mutations contributing to epilepsy syndromes and the modulation of autophagy playing a role in seizure onset. The apoptotic pathway contribute to cell death through glutamate receptor-mediated excitotoxicity, involving pro-apoptotic proteins like p53 and mitochondrial dysfunction, leading to the activation of caspases and the disruption of calcium homeostasis. Ionic imbalances within neural networks contribute to the complexity of epileptic seizures, involving alterations in voltage-gated sodium and potassium channels, and the formation of diverse ion channel subtypes. Epileptogenesis triggers molecular changes in hippocampus, including altered neurogenesis and enhanced expression of neurotrophic factors and proteins. Oxidative stress leads to cellular damage, disrupted antioxidant systems, and mitochondrial dysfunction, making it a key player in epileptogenesis and potential neuroprotective interventions. Thalamocortical circuitry disruption is central to absence epilepsy, the normal circuit becomes faulty and results in characteristic brain wave patterns.
Keywords: Epilectic seizures; Epilepsy; Epileptogenesis; Molecular mechanism.
© 2023. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Ethics approval and consent to participate is not applicable in this study. Consent for publication: Consent for publication is not applicable in this study. Competing interests: All authors declare that there is no competing interests.
Figures
Similar articles
-
Multiple Disruptions of Glial-Neuronal Networks in Epileptogenesis That Follows Prolonged Febrile Seizures.Front Neurol. 2021 Feb 18;12:615802. doi: 10.3389/fneur.2021.615802. eCollection 2021. Front Neurol. 2021. PMID: 33679583 Free PMC article.
-
Pharmacological modulation of cytokines correlating neuroinflammatory cascades in epileptogenesis.Mol Biol Rep. 2022 Feb;49(2):1437-1452. doi: 10.1007/s11033-021-06896-8. Epub 2021 Nov 9. Mol Biol Rep. 2022. PMID: 34751915 Review.
-
Epilepsy and Alterations of the Blood-Brain Barrier: Cause or Consequence of Epileptic Seizures or Both?Handb Exp Pharmacol. 2022;273:331-350. doi: 10.1007/164_2020_406. Handb Exp Pharmacol. 2022. PMID: 33136189
-
Neuronal injury in chronic CNS inflammation.Best Pract Res Clin Anaesthesiol. 2010 Dec;24(4):551-62. doi: 10.1016/j.bpa.2010.11.001. Epub 2010 Nov 29. Best Pract Res Clin Anaesthesiol. 2010. PMID: 21619866 Review.
-
An Insight into Molecular Mechanisms and Novel Therapeutic Approaches in Epileptogenesis.CNS Neurol Disord Drug Targets. 2020;19(10):750-779. doi: 10.2174/1871527319666200910153827. CNS Neurol Disord Drug Targets. 2020. PMID: 32914725
Cited by
-
The role of L-DOPA in neurological and neurodegenerative complications: a review.Mol Cell Biochem. 2025 Jun 9. doi: 10.1007/s11010-025-05324-w. Online ahead of print. Mol Cell Biochem. 2025. PMID: 40488810 Review.
-
Treatment outcomes, medication adherence and predictors among patients with epilepsy in Mekelle City Hospitals, Ethiopia: a multicentre observational cross-sectional study.BMJ Open. 2025 Jun 24;15(6):e097067. doi: 10.1136/bmjopen-2024-097067. BMJ Open. 2025. PMID: 40555444 Free PMC article.
-
Role of hippocampus in epileptogenesis: new insights in the cross-talks between the underlying mechanisms.Acta Neurol Belg. 2025 Aug 9. doi: 10.1007/s13760-025-02857-1. Online ahead of print. Acta Neurol Belg. 2025. PMID: 40783473 Review.
-
Unraveling the nutritional challenges in epilepsy: Risks, deficiencies, and management strategies: A systematic review.World J Exp Med. 2025 Jun 20;15(2):104328. doi: 10.5493/wjem.v15.i2.104328. eCollection 2025 Jun 20. World J Exp Med. 2025. PMID: 40546663 Free PMC article.
-
In Silico molecular docking and molecular dynamic simulation of transferrin coated Phenytoin loaded SLNs with molecular targets of epilepsy.PLoS One. 2025 Jun 20;20(6):e0325772. doi: 10.1371/journal.pone.0325772. eCollection 2025. PLoS One. 2025. PMID: 40540445 Free PMC article.
References
-
- Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, Elger CE, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55(4):475–82. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous