

Genetic variant reanalysis reveals a case of Sandhoff disease with onset of infantile epileptic spasm syndrome
- PMID: 40217552
- PMCID: PMC11960328
- DOI: 10.1186/s42494-024-00149-4
Genetic variant reanalysis reveals a case of Sandhoff disease with onset of infantile epileptic spasm syndrome
Abstract
Background: Sandhoff disease (SD) i s an autosomal recessive lysosomal disease with clinical manifestations such as epilepsy, psychomotor retardation and developmental delay. However, infantile SD with onset of infantile epilepsy spasm syndrome (IESS) is extremely rare.
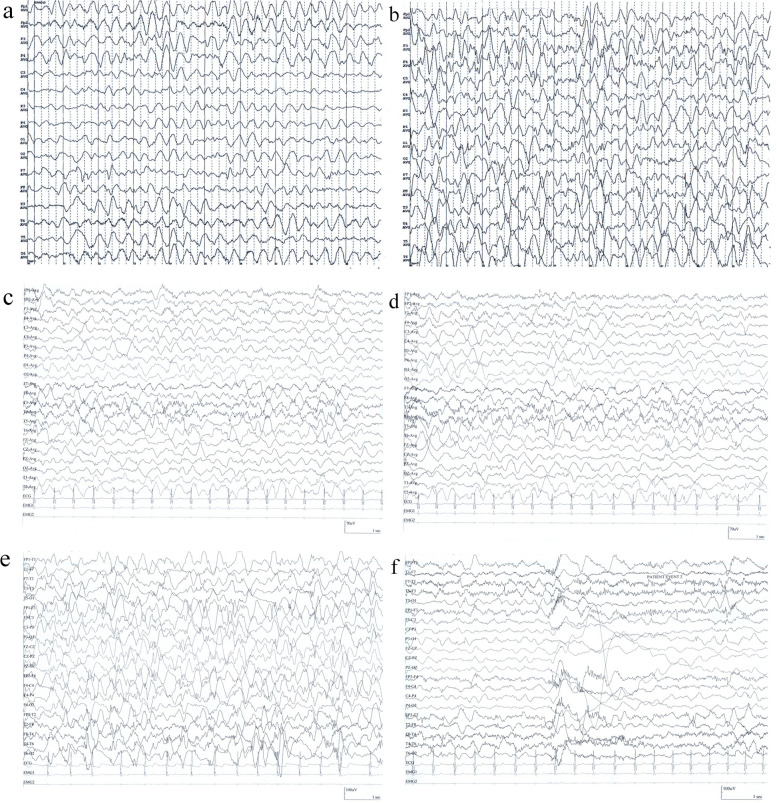
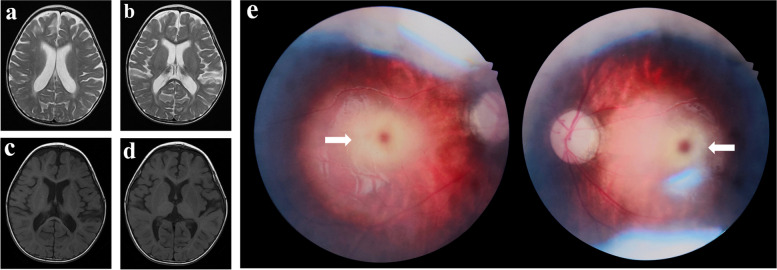
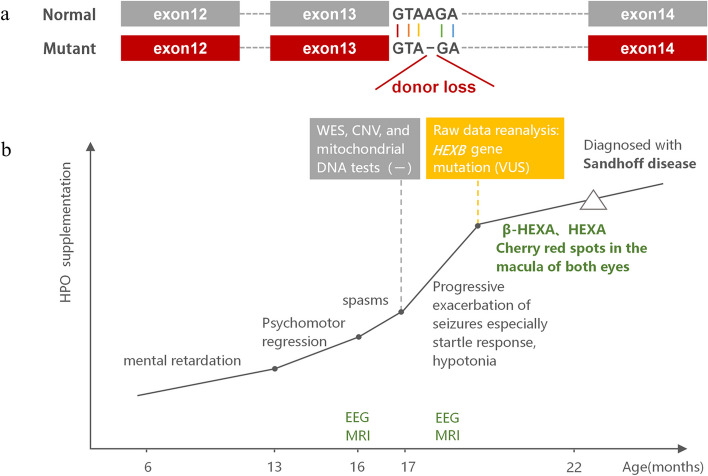
Case presentation: The case presented here was a 22-month-old boy, who presented with IESS and psychomotor retardation/regression at 6 months of age. The patient showed progressive aggravation of seizures and excessive startle responses. The whole exome sequencing data, which initially revealed negative results, were reanalyzed and indicated a homozygous mutation at the c.1613 + 4del splice site of the HEXB gene. The activities of β-hexosaminidase A and total hexosaminidase were significantly decreased. The fundus examination showed cherry red spots at the macula.
Conclusions: IESS can be an epileptic phenotype of infantile SD. Clinical phenotypes should be adequately collected in genetic testing. In the case of negative sequencing results, gene variant reanalysis can be performed when the patients show clinically suspicious indications.
Keywords: HEXB gene; Cherry red spot; Gene variant reanalysis; Human phenotype ontology; Infantile Sandhoff disease; Infantile epilepsy spasm syndrome.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: The studies involving human participants were reviewed and approved by the the Institutional Review Board of Chinese PLA General Hospital(The ethics number: 2023–143). Written informed consent to participate in this study was provided by the participants’ legal guardian. Competing interests: Author Liping Zou is the member of the Editorial Board for Acta Epileptologica, who was not involved in the journal’s review of, or decisions related to this manuscript.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous