

Understanding the Natural History and the Effects of Current Therapeutic Strategies on Urea Cycle Disorders: Insights from the UCD Spanish Registry
- PMID: 40218931
- PMCID: PMC11990916
- DOI: 10.3390/nu17071173
Understanding the Natural History and the Effects of Current Therapeutic Strategies on Urea Cycle Disorders: Insights from the UCD Spanish Registry
Abstract
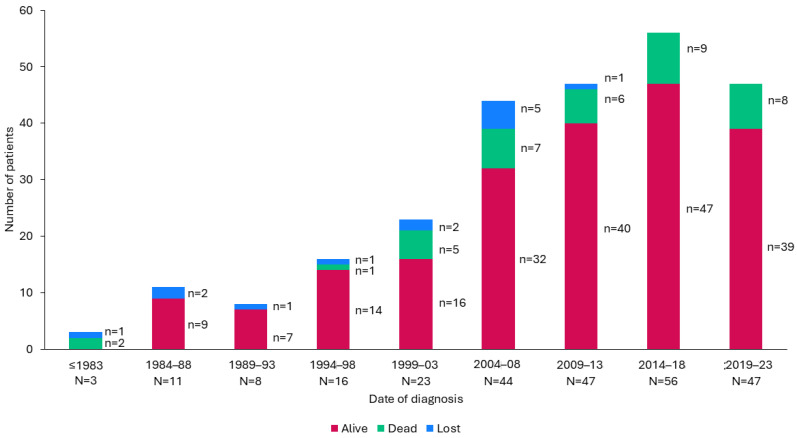
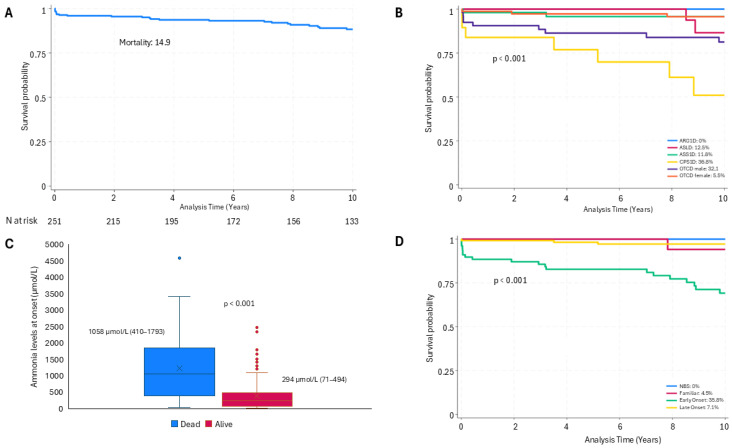
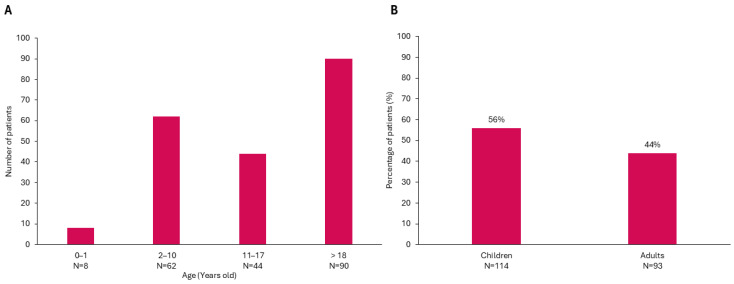
Background/Objectives: The present study updates the Spanish registry of patients with urea cycle disorders (UCD), originally established in 2013, to provide comprehensive epidemiological data and evaluate the impact of therapeutic strategies and newborn screening (NBS) on clinical outcomes. Methods: This retrospective, multicenter study focuses on 255 Spanish UCD patients. It includes all living and deceased cases up to February 2024, analyzing demographic, clinical, and biochemical variables. Results: The incidence of UCD in Spain over the past decade was 1:36,063 births. The most common defects were ornithine transcarbamylase deficiency (OTCD) and argininosuccinate synthetase deficiency. Early-onset (EO) cases comprised 32.7%, and 10.6% were diagnosed through NBS. Global mortality was 14.9%, higher in carbamoylphosphate synthetase 1 deficiency (36.8%) and male OTCD patients (32.1%) compared to other defects (p = 0.013). EO cases presented a higher mortality rate (35.8%) than late-onset (LO) cases (7.1%) (p < 0.0001). The median ammonia level in deceased patients was higher at 1058 µmol/L (IQR 410-1793) than in survivors at 294 µmol/L (IQR 71-494) (p < 0.0001). Diagnosis through NBS improved survival and reduced neurological impairment compared to symptomatic diagnosis. Neurological impairment occurred in 44% of patients, with worse neurological outcomes observed in patients with argininosuccinate lyase deficiency, arginase 1 deficiency, hyperornithinemia-hyperammonemia-homocitrullinuria, EO presentations, pre-2014 diagnosis, and patients with higher levels of ammonia at diagnosis. Among transplanted patients (20.6%), survival was 95.2%, with no significant neurological differences compared to non-transplanted patients. Conclusions: This updated analysis highlights the positive impact of NBS and advanced treatments on mortality and neurologic outcomes. Persistent neurological challenges underscore the need for further therapeutic strategies.
Keywords: N-acetylglutamate synthase (NAGS); arginase 1 (ARG1); argininosuccinate lyase (ASL); argininosuccinate synthetase (ASS1); carbamoylphosphate synthetase (CPS1); carbonic anhydrase VA (CA-VA); citrin; hyperornithinemia-hyperammonemia-homocitrullinuria (HHH); ornithine transcarbamylase (OTC); ornithine/citrulline antiporter (ORNT); urea cycle disorders (UCDs).
Conflict of interest statement
E.M.-H. has received financial support for attending conferences from Sanofi Spain, Lucane, and Nutricia Danone and has received writing support from Immedica Pharma Spain. None of them have influenced the content of this manuscript, and therefore, no conflicts of interest are declared. E.C.-S. received honoraria from Takeda as a speaker, received reimbursements from Lucane Pharma, Biomarin Pharmaceutical, Eisai and Takeda for attending scientific meetings and medical congresses, but E.C. confirms independence from the sponsors and that the content of the article has not been influenced by them. A.B.-Q. has received speaker fees, meeting funding, advisory work, or investigation grants from various companies (Danone, Nestle, Recordati Rare Diseases, Grand Fontaine, Rendon, Sobi, Sanofi, Shire, Takeda, Biomarin, PTC, Jnana) over the last 5 years. None of these have influenced the content of this manuscript, and therefore, no conflicts of interest are declared. S.S. has received speaker and travel fees from Sanofi Spain, Nutricia Danone and Nestlé Vitaflo. A.G.-C. has received honoraria for research support and lectures from PTC Therapeutics and Immedica, honoraria for lectures from Biomarin, Immedica, Eisai, Orchard Therapeutics, and Recordati Rare Diseases Foundation, and is a co-founder of the Hospital Sant Joan de Déu start-up ‘Neuroprotect Life Sciences’. M.B. has received financial support for attending conferences and/or giving lectures from Lucane Pharma, Nutricia Metabolics, and Orphan Europe. Á.M.-R. and M.M.H. have received writing support from Immedica Pharma Spain. B.P. has received financial support for attending conferences or honoraria from PTC Therapeutics, Chiesi, and Immedica Pharma Spain. None of these entities have influenced the content of this manuscript, and therefore, no conflicts of interest are declared. The rest of the authors declare no conflict of interest with this study. The sponsors had no role in the design, execution, interpretation, or writing of the study.
Figures
References
-
- Häberle J., Burlina A., Chakrapani A., Dixon M., Karall D., Lindner M., Mandel H., Martinelli D., Pintos-Morell G., Santer R., et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. J. Inherit. Metab. Dis. 2019;42:1192–1230. doi: 10.1002/jimd.12100. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
