

Diagnosis of a patient with severe sensorineural hearing loss as the initial symptom caused by novel compound heterozygous variant in SLC19A2 gene
- PMID: 40220483
- PMCID: PMC12017973
- DOI: 10.1016/j.bjorl.2025.101581
Diagnosis of a patient with severe sensorineural hearing loss as the initial symptom caused by novel compound heterozygous variant in SLC19A2 gene
Abstract
Objective: Thiamine-Responsive Megaloblastic Anemia (TRMA) syndrome, caused by biallelic variants in the SLC19A2 gene, typically presents with a triad of megaloblastic anemia, diabetes mellitus, and sensorineural hearing loss. This study aims to determine the genetic etiology and clinical phenotype of a patient who presented with severe sensorineural hearing loss as the initial symptom, and to expand our understanding of the SLC19A2 variant spectrum.
Methods: Proband-only whole-exome sequencing was performed to screen the candidate variants, which were subsequently validated by Sanger sequencing within the family. cDNA sequencing based on RT-PCR and TA cloning analysis was used to determine the effect of splicing variants on mRNA processing of SLC19A2 gene. Detailed clinical features were evaluated by a diagnostic hearing test, laboratory and imaging examination.
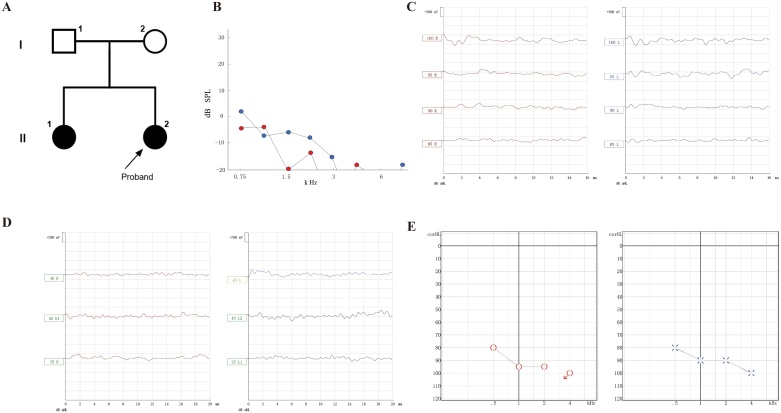
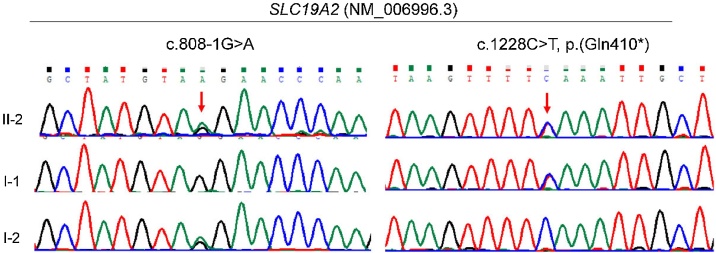
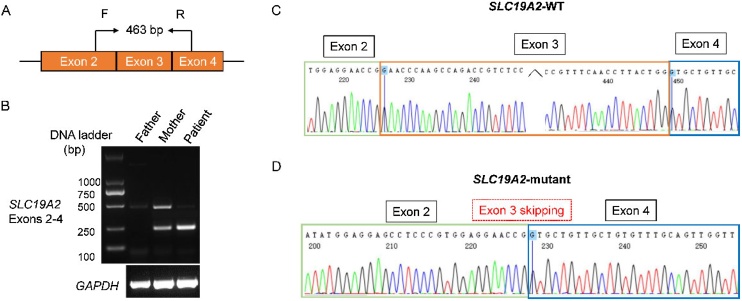
Results: A 2-year-5-month-old Chinese girl was diagnosed with diabetes mellitus and severe sensorineural hearing loss, without abnormal hemoglobin. DNA sequencing revealed a novel compound heterozygous variant of c.808-1G > A and c.1228C > T (p.Gln410*) in the SLC19A2 gene. Both variants were previously unreported. The c.808-1G > A splicing variant is located in intron 2 of SLC19A2, and is predicted to cause exon 3 skipping. The cDNA experiment confirmed this biological event, further indicating that the splicing variant can cause amino acid frameshift alteration (p.Glu270Valfs*10) in SLC19A2.
Conclusion: We report a patient with TRMA syndrome (without anemia) caused by a novel compound heterozygous variant in SLC19A2 gene. This study suggests that the possibility of TRMA syndrome should be considered when encountering patients with early-onset severe sensorineural hearing loss in clinical practice.
Level of evidence: Level 4.
Keywords: Functional study; Novel compound heterozygous variant; SLC19A2 gene; Sensorineural hearing loss; Thiamine-responsive megaloblastic anemia syndrome.
Copyright © 2025. Published by Elsevier España S.L.U.
Conflict of interest statement
Declaration of competing interest The authors declare no conflicts of interest.
Figures
Similar articles
-
Thiamine-Responsive Megaloblastic Anemia Syndrome Mimicking Myelodysplastic Neoplasm.Acta Haematol. 2025;148(4):380-385. doi: 10.1159/000542286. Epub 2024 Oct 28. Acta Haematol. 2025. PMID: 39467528 Free PMC article.
-
Thiamine responsive megaloblastic anemia: a novel SLC19A2 compound heterozygous mutation in two siblings.Pediatr Diabetes. 2013 Aug;14(5):384-7. doi: 10.1111/j.1399-5448.2012.00921.x. Epub 2013 Jan 4. Pediatr Diabetes. 2013. PMID: 23289844
-
Leber's congenital amaurosis as the retinal degenerative phenotype in thiamine responsive megaloblastic anemia: a case report.Ophthalmic Genet. 2014 Jun;35(2):119-24. doi: 10.3109/13816810.2013.793363. Epub 2013 May 2. Ophthalmic Genet. 2014. PMID: 23638917
-
Sickle Cell Disease.2003 Sep 15 [updated 2025 Feb 13]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2003 Sep 15 [updated 2025 Feb 13]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 20301551 Free Books & Documents. Review.
-
Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19.Cochrane Database Syst Rev. 2022 May 20;5(5):CD013665. doi: 10.1002/14651858.CD013665.pub3. Cochrane Database Syst Rev. 2022. PMID: 35593186 Free PMC article.
References
-
- Labay V., Raz T., Baron D., et al. Mutations in SLC19A2 cause thiamine-responsive megaloblastic anaemia associated with diabetes mellitus and deafness. Nat Genet. 1999;22:300–304. - PubMed
-
- Ortigoza-Escobar J.D., Molero-Luis M., Arias A., et al. Free-thiamine is a potential biomarker of thiamine transporter-2 deficiency: a treatable cause of Leigh syndrome. Brain. 2016;139:31–38. - PubMed
-
- Marce-Grau A., Marti-Sanchez L., Baide-Mairena H., Ortigoza-Escobar J.D., Perez-Duenas B. Genetic defects of thiamine transport and metabolism: a review of clinical phenotypes, genetics, and functional studies. J Inherit Metab Dis. 2019;42:581–597. - PubMed
-
- Sako S., Tsunogai T., Oishi K., et al. In: Thiamine-responsive megaloblastic anemia syndrome. Adam M.P., Feldman J., Mirzaa G.M., editors. GeneReviews((R)); Seattle (WA): 1993. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Research Materials
