

Regulation of nucleotide excision repair by wild-type HRAS signaling in head and neck cancer
- PMID: 40221503
- PMCID: PMC12183087
- DOI: 10.1038/s41417-025-00902-y
Regulation of nucleotide excision repair by wild-type HRAS signaling in head and neck cancer
Abstract
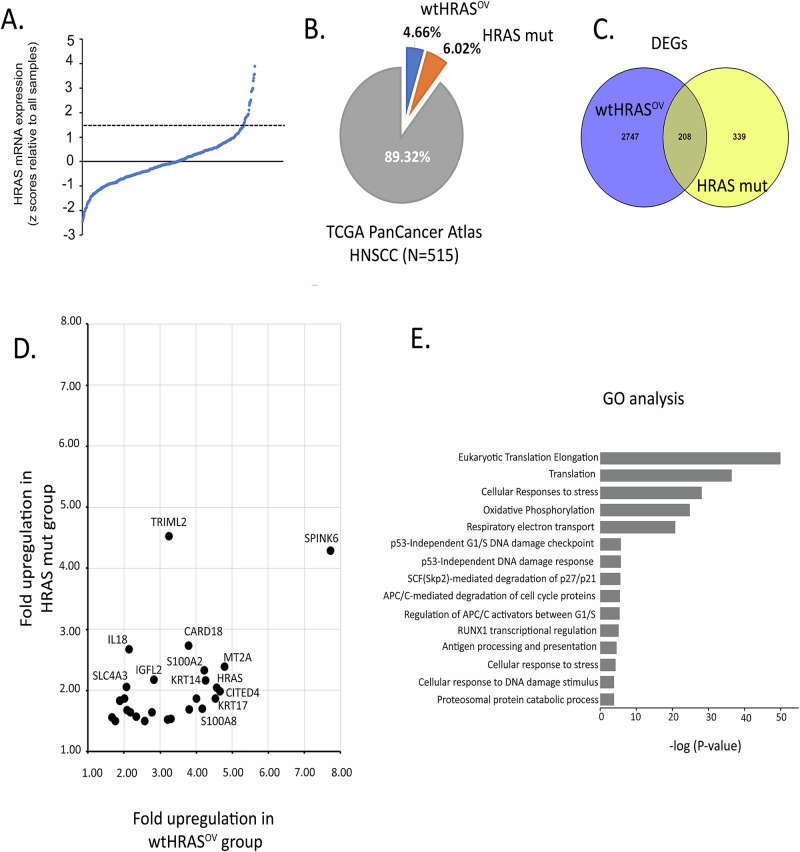
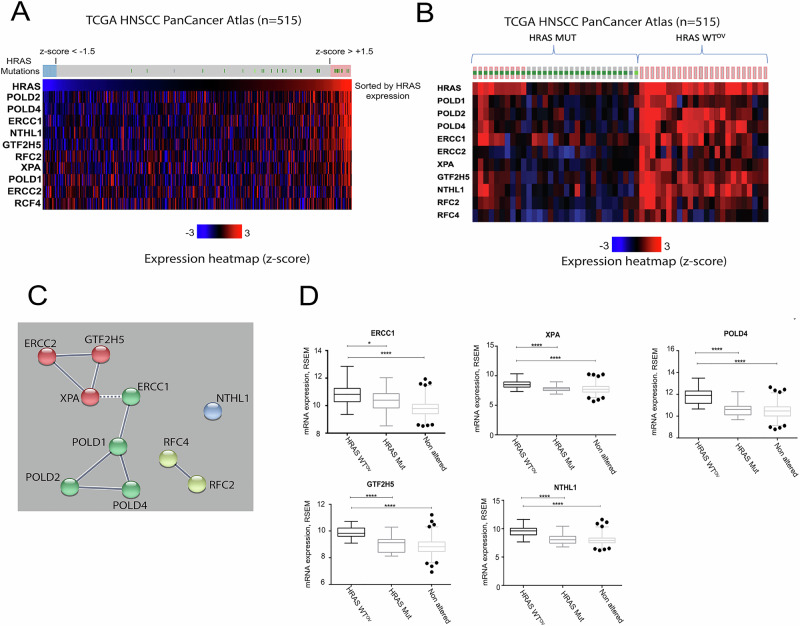
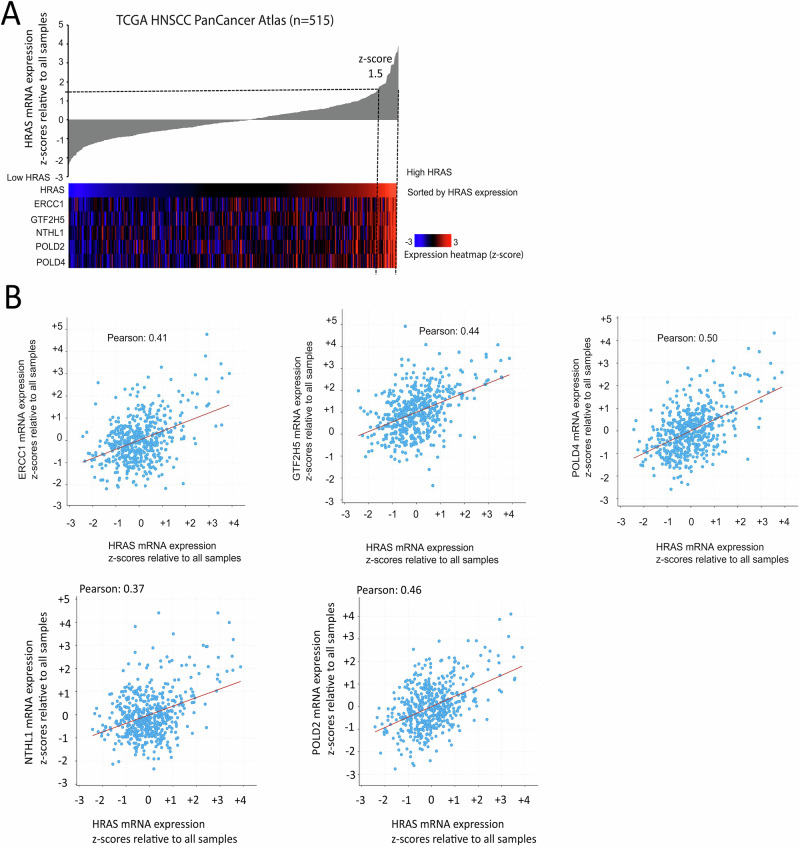
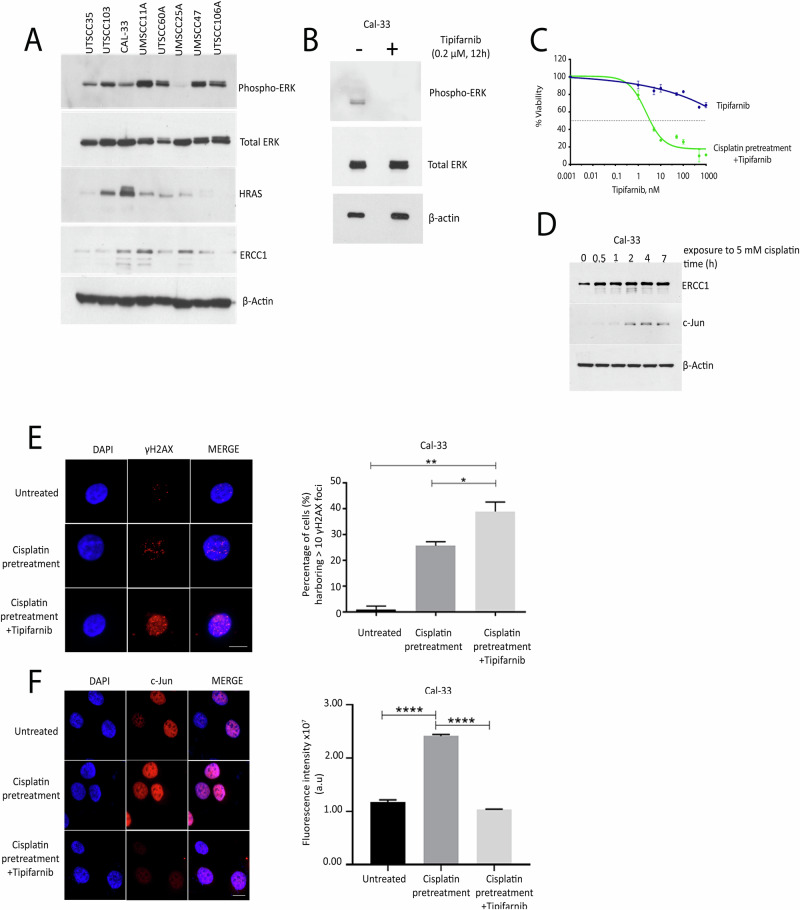
Head and neck squamous cell carcinoma (HNSCC) is characterized by a high rate of locoregional or distant relapse among patients. It is well established that resistance to chemotherapeutic drugs has an important role in the emergence of the recurrent and/or metastatic type of this malignancy which is associated with poor prognosis. Therefore, understanding the molecular basis of chemoresistance in head and neck cancer is required for the development of effective therapeutic strategies. Activating mutations in the HRAS gene are driver events in human cancer. Although numerous studies have demonstrated that oncogenic HRAS mutations promote chemoresistance in HNSCC, the molecular profile of HNSCC tumors that overexpress wild-type HRAS (wtHRASov) and their response to chemotherapy is poorly investigated. To gain deeper insights into the characteristics of wtHRASov tumors, we conducted a gene expression analysis using transcriptome data from The Cancer Genome Atlas (TCGA). This analysis revealed a distinct signature of overexpressed nucleotide excision repair (NER) genes in wtHRASov tumors, which are associated with chemoresistance. We further explored the role of these NER components in response to genotoxic stress, utilizing a diverse panel of HNSCC cell lines and patient-derived xenografts. Our findings indicate that in a specific cluster of head and neck tumors, ERK/cJun signaling activation is strongly reliant on HRAS activity. Inhibiting HRAS in these tumors results in a significant downregulation of the NER signature components, re-sensitizing cancer cells to platinum-based chemotherapy.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: TR reports research funding from Kura Oncology through a sponsored research contract. The remaining authors declare no conflict of interest. Ethics approval: Tumor samples used for patient-derived xenografts were obtained from patients who underwent laryngeal or oral cancer surgery at the 2nd Department of Otolaryngology, National and Kapodistrian University of Athens, University General Hospital “Attikon,” between 2018 and 2019. This study adhered to the ethical standards of the 1975 Declaration of Helsinki (revised in 2008) and was approved (07.01.2009/4256) and renewed (521/20.7.18) by the Ethics Committee of the National and Kapodistrian University of Athens. All patients provided informed consent. All mice experiments were performed in the animal facility of BRFAA (Biomedical Research Foundation of the Academy of Athens) according to national and international regulations and were approved by the BRFAA ethical committee.
Figures
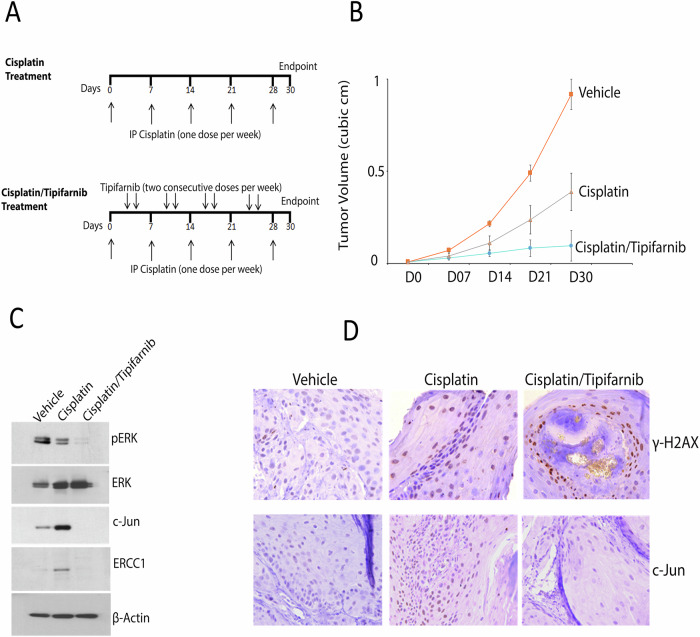
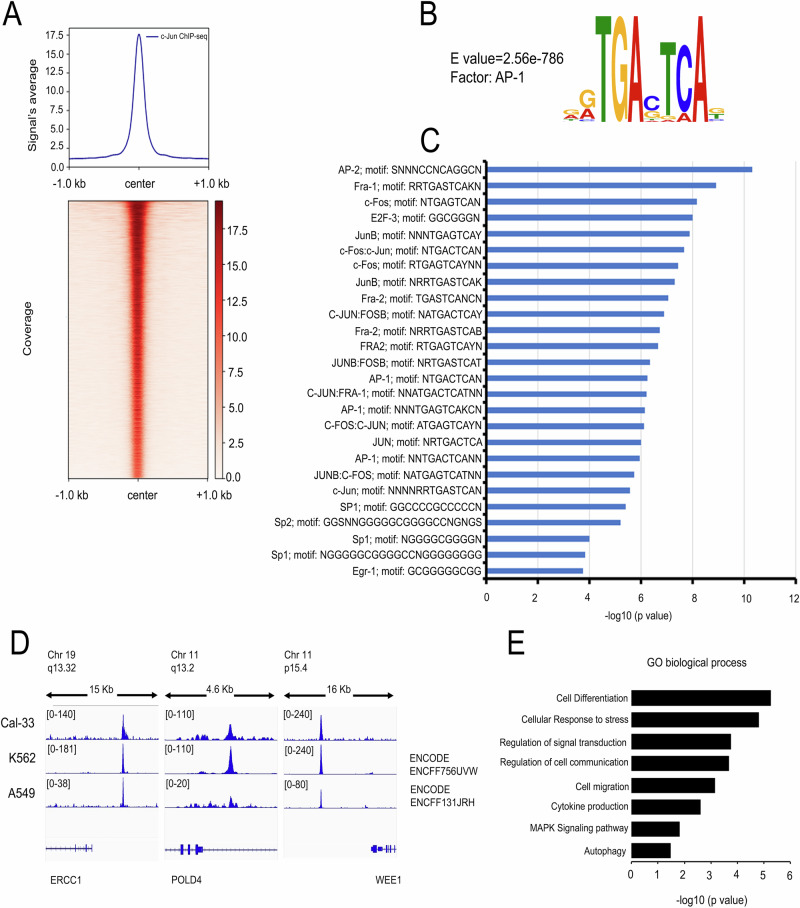
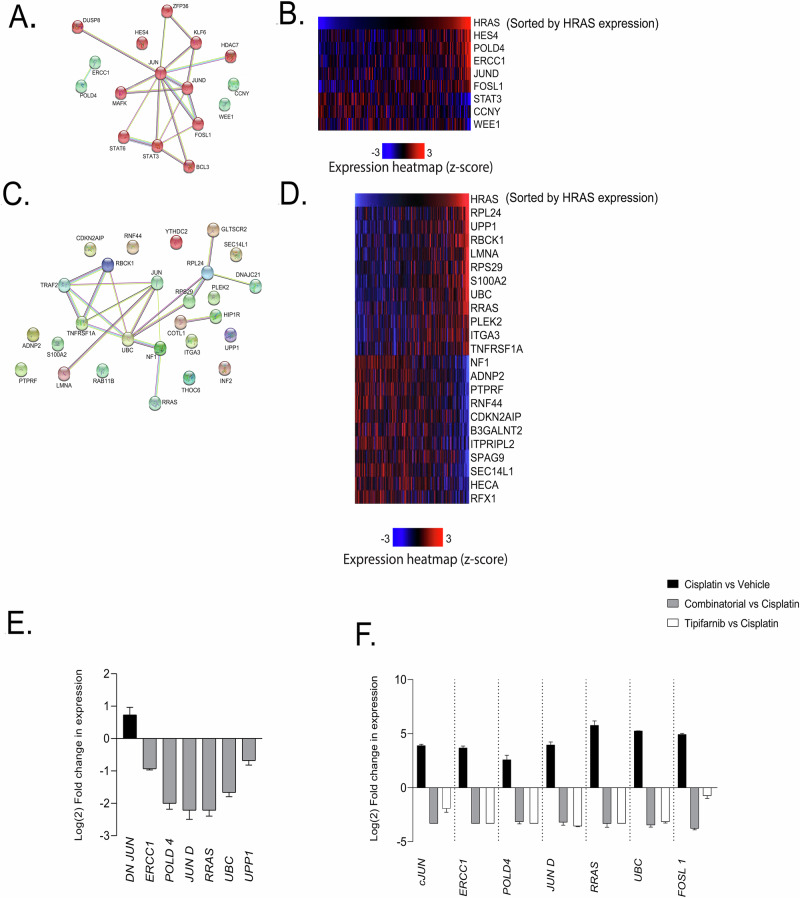
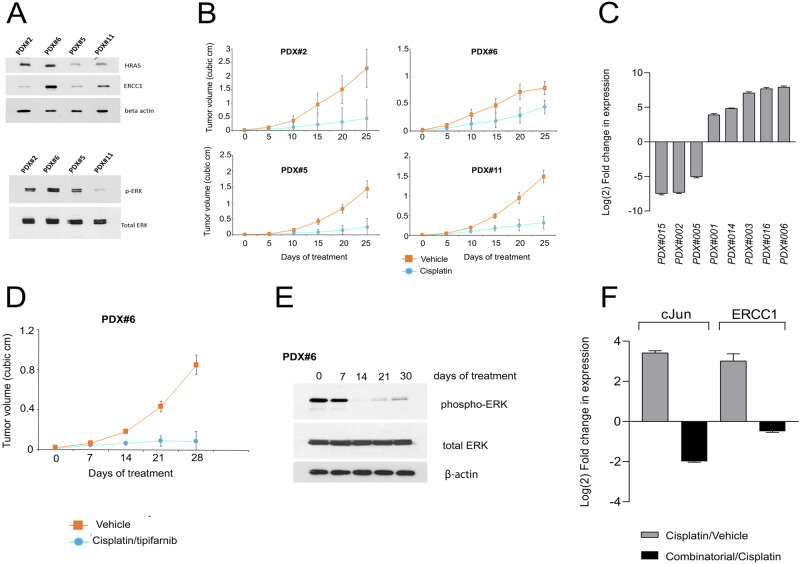
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
