

Deep learning tools predict variants in disordered regions with lower sensitivity
- PMID: 40221640
- PMCID: PMC11992697
- DOI: 10.1186/s12864-025-11534-9
Deep learning tools predict variants in disordered regions with lower sensitivity
Abstract
Background: The recent AI breakthrough of AlphaFold2 has revolutionized 3D protein structural modeling, proving crucial for protein design and variant effects prediction. However, intrinsically disordered regions-known for their lack of well-defined structure and lower sequence conservation-often yield low-confidence models. The latest Variant Effect Predictor (VEP), AlphaMissense, leverages AlphaFold2 models, achieving over 90% sensitivity and specificity in predicting variant effects. However, the effectiveness of tools for variants in disordered regions, which account for 30% of the human proteome, remains unclear.
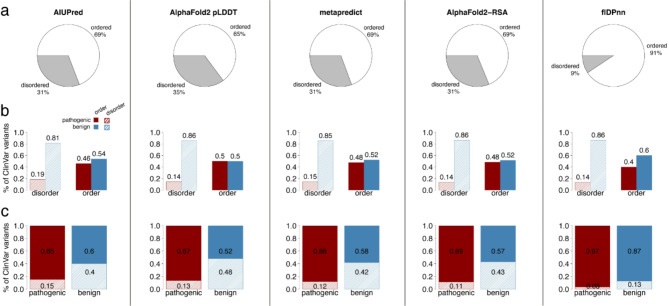
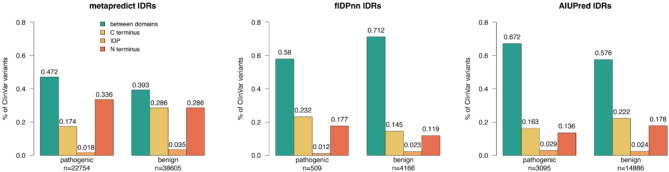
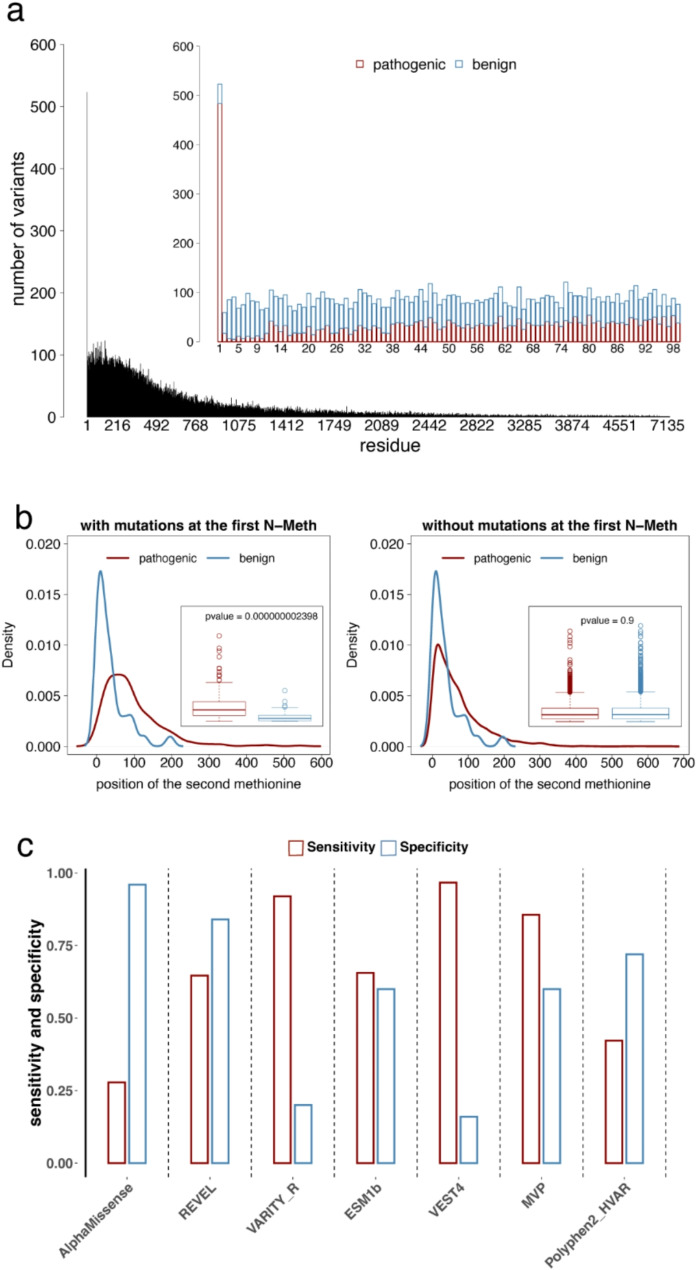
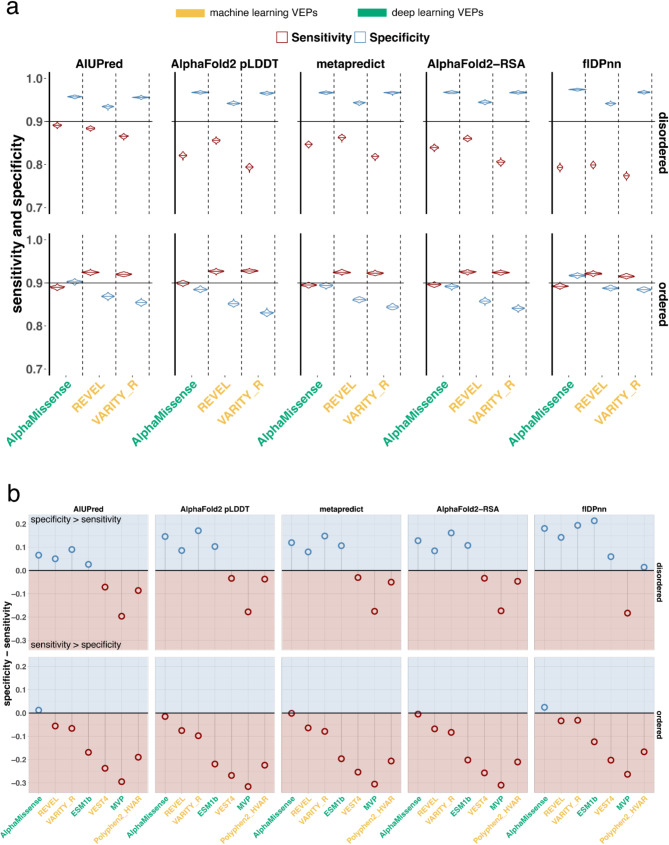
Results: In this study, we found that predicting pathogenicity for variants in disordered regions is less accurate than in ordered regions, particularly for mutations at the first N-Methionine site. Investigations into the efficacy of variant effect predictors on intrinsically disordered regions (IDRs) indicated that mutations in IDRs are predicted with lower sensitivity and the gap between sensitivity and specificity is largest in disordered regions, especially for AlphaMissense and VARITY.
Conclusions: The prevalence of IDRs within the human proteome, coupled with the increasing repertoire of biological functions they are known to perform, necessitated an investigation into the efficacy of state-of-the-art VEPs on such regions. This analysis revealed their consistently reduced sensitivity and differing prediction performance profile to ordered regions, indicating that new IDR-specific features and paradigms are needed to accurately classify disease mutations within those regions.
Keywords: AlphaMissense; Benchmarking; Intrinsically disordered regions; Methionine start site; Variant effect predictors.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: N/A. Consent for publication: N/A. Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
AlphaFold and Implications for Intrinsically Disordered Proteins.J Mol Biol. 2021 Oct 1;433(20):167208. doi: 10.1016/j.jmb.2021.167208. Epub 2021 Aug 18. J Mol Biol. 2021. PMID: 34418423 Review.
-
A Comprehensive Survey of the Roles of Highly Disordered Proteins in Type 2 Diabetes.Int J Mol Sci. 2017 Sep 21;18(10):2010. doi: 10.3390/ijms18102010. Int J Mol Sci. 2017. PMID: 28934129 Free PMC article.
-
Conformational ensembles of the human intrinsically disordered proteome.Nature. 2024 Feb;626(8000):897-904. doi: 10.1038/s41586-023-07004-5. Epub 2024 Jan 31. Nature. 2024. PMID: 38297118
-
Insights into the evolutionary forces that shape the codon usage in the viral genome segments encoding intrinsically disordered protein regions.Brief Bioinform. 2021 Sep 2;22(5):bbab145. doi: 10.1093/bib/bbab145. Brief Bioinform. 2021. PMID: 33866372
-
Towards Decoding the Sequence-Based Grammar Governing the Functions of Intrinsically Disordered Protein Regions.J Mol Biol. 2021 Jun 11;433(12):166724. doi: 10.1016/j.jmb.2020.11.023. Epub 2020 Nov 26. J Mol Biol. 2021. PMID: 33248138 Review.
Cited by
-
Molecular dynamics simulations of intrinsically disordered protein regions enable biophysical interpretation of variant effect predictors.bioRxiv [Preprint]. 2025 May 12:2025.05.07.652723. doi: 10.1101/2025.05.07.652723. bioRxiv. 2025. PMID: 40462930 Free PMC article. Preprint.
References
-
- Pentony MM, Jones DT. Modularity of intrinsic disorder in the human proteome: disorder in the human proteome. Proteins. 2010;78:212–21. - PubMed
-
- Ruff KM, Pappu RV. AlphaFold and implications for intrinsically disordered proteins. J Mol Biol. 2021;433:167208. - PubMed
-
- Tesei G, Trolle AI, Jonsson N, Betz J, Knudsen FE, Pesce F, et al. Conformational ensembles of the human intrinsically disordered proteome. Nature. 2024. 10.1038/s41586-023-07004-5. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
