

What is the phenotype of heterozygous lipoprotein lipase deficiency?
- PMID: 40223670
- PMCID: PMC11888829
- DOI: 10.1097/MOL.0000000000000974
What is the phenotype of heterozygous lipoprotein lipase deficiency?
Abstract
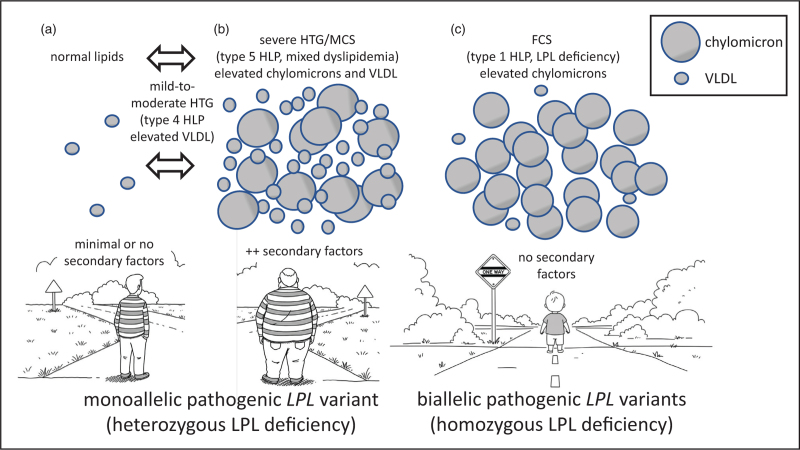
Purpose of review: Genetic testing of patients with severe hypertriglyceridemia often identifies a single heterozygous pathogenic variant in the LPL gene. The complex and variable phenotype associated with this genotype is the topic of this review.
Recent findings: Previous research showed that heterozygosity for lipoprotein lipase deficiency is associated with reduced but variable post heparin lipolytic activity alongside inconsistent plasma lipid phenotypes ranging from normal to mild-to-moderate to severe hypertriglyceridemia. Recent research confirms and extends these observations, showing that a heterozygous individual can express a highly variable phenotype over time, depending on the presence of secondary factors. About 10% (range 8-20%) of patients with severe hypertriglyceridemia or multifactorial chylomicronemia syndrome are heterozygous for a rare pathogenic LPL variant, and a clinically relevant minority of these has recalcitrant or sustained hypertriglyceridemia.
Summary: Heterozygosity for lipoprotein lipase deficiency predisposes to hypertriglyceridemia, which is sometimes severe depending on secondary factors, but is typically quite responsive to routine interventions such as diet, lifestyle and existing lipid-lowering therapies. However, many heterozygotes for pathogenic variants in LPL have completely normal plasma lipids.
Keywords: chylomicronemia; familial chylomicronemia syndrome; hyperlipoproteinemia type 1; hyperlipoproteinemia type 4; hyperlipoproteinemia type 5; hypertriglyceridemia; lipoprotein lipase; lipoprotein lipase deficiency; multifactorial chylomicronemia syndrome; pathogenic variant.
Copyright © 2025 The Author(s). Published by Wolters Kluwer Health, Inc.
Conflict of interest statement
Figures

Similar articles
-
The longitudinal triglyceride phenotype in heterozygotes with LPL pathogenic variants.J Clin Lipidol. 2023 Jan-Feb;17(1):87-93. doi: 10.1016/j.jacl.2022.11.007. Epub 2022 Nov 22. J Clin Lipidol. 2023. PMID: 36476373
-
Molecular analysis of three known and one novel LPL variants in patients with type I hyperlipoproteinemia.Nutr Metab Cardiovasc Dis. 2018 Feb;28(2):158-164. doi: 10.1016/j.numecd.2017.11.003. Epub 2017 Nov 22. Nutr Metab Cardiovasc Dis. 2018. PMID: 29288010
-
Incidental finding of severe hypertriglyceridemia in children. Role of multiple rare variants in genes affecting plasma triglyceride.J Clin Lipidol. 2017 Nov-Dec;11(6):1329-1337.e3. doi: 10.1016/j.jacl.2017.08.017. Epub 2017 Sep 4. J Clin Lipidol. 2017. PMID: 28951076
-
Lipoprotein Lipase: Structure, Function, and Genetic Variation.Genes (Basel). 2025 Jan 5;16(1):55. doi: 10.3390/genes16010055. Genes (Basel). 2025. PMID: 39858602 Free PMC article. Review.
-
Identification of a Compound Heterozygous LMF1 Variants in a Patient with Severe Hypertriglyceridemia - Case Report and Literature Review.J Atheroscler Thromb. 2024 Jul 1;31(7):1106-1111. doi: 10.5551/jat.64697. Epub 2024 Mar 9. J Atheroscler Thromb. 2024. PMID: 38462482 Free PMC article. Review.
References
-
- Brown EE, Sturm AC, Cuchel M, et al. . Genetic testing in dyslipidemia: a scientific statement from the National Lipid Association. J Clin Lipidol 2020; 14:398–413. - PubMed
-
- Berberich AJ, Hegele RA. The complex molecular genetics of familial hypercholesterolaemia. Nat Rev Cardiol 2019; 16:9–20. - PubMed
-
- Lazarte J, Hegele RA. Can genetic testing help in the management of dyslipidaemias? Curr Opin Lipidol 2020; 31:187–193. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
