

Injured tubular epithelia-derived CCN1 promotes the mobilization of fibroblasts toward injury sites after kidney injury
- PMID: 40224016
- PMCID: PMC11987671
- DOI: 10.1016/j.isci.2025.112176
Injured tubular epithelia-derived CCN1 promotes the mobilization of fibroblasts toward injury sites after kidney injury
Abstract
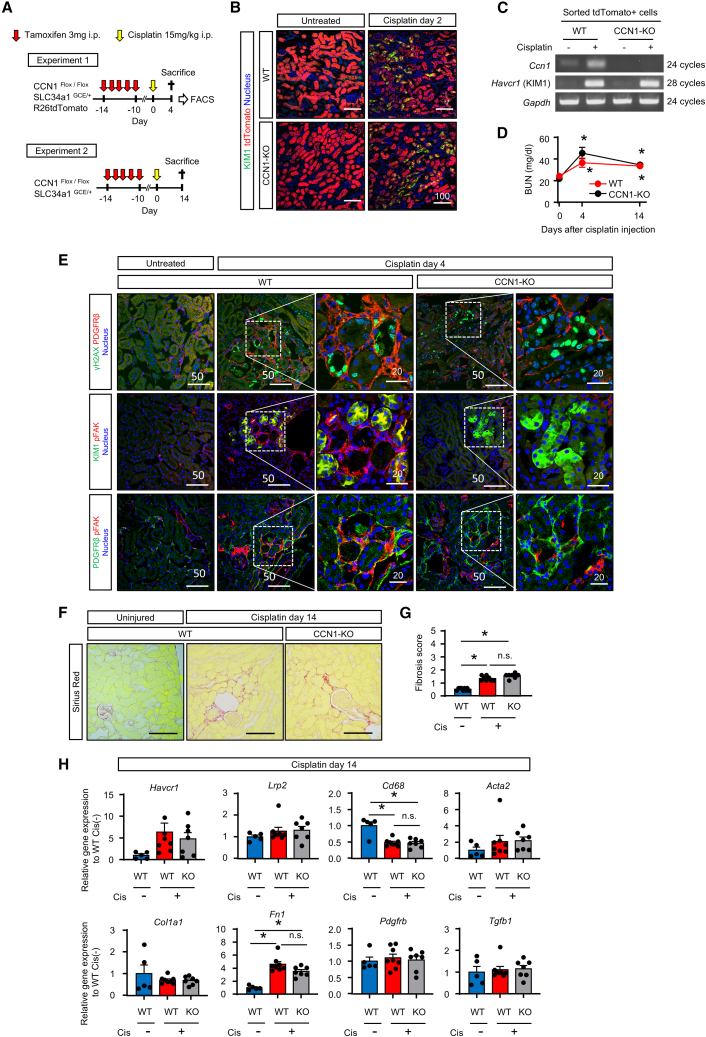
Humoral factors that prompt fibroblasts to migrate to an injury site at an appropriate time point are deemed indispensable for repair after kidney injury. We herein demonstrated the pivotal roles of injured tubule-derived cellular communication network factor 1 (CCN1) in the mobilization of fibroblasts to the injury site after kidney injury. Based on analyses of ligand-receptor interactions in vitro and tubular epithelial-specific transcriptomics in vivo, we identified the up-regulation of CCN1 during the early phases of kidney injury. CCN1 promotes fibroblast chemotaxis through focal adhesion kinase-extracellular signal-regulated kinase (ERK) signaling. In vivo analyses utilizing tubular-specific CCN1 knockout (KO) mice demonstrated the sparse accumulation of fibroblasts around injured sites after injury, resulting in ameliorated tissue fibrosis in CCN1-KO mice. These results reveal an epithelial-fibroblast CCN1 signaling axis that mobilizes fibroblasts to injured tubule early after acute injury but that promotes interstitial fibrosis at late time points.
Keywords: biochemical mechanism; biology of human development; integrative aspects of cell biology; molecular interaction.
© 2025 The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures







References
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
