

The Common PKD1 p.(Ile3167Phe) Variant Is Hypomorphic and Associated with Very Early Onset, Biallelic Polycystic Kidney Disease
- PMID: 40225160
- PMCID: PMC11918491
- DOI: 10.1155/2023/5597005
The Common PKD1 p.(Ile3167Phe) Variant Is Hypomorphic and Associated with Very Early Onset, Biallelic Polycystic Kidney Disease
Abstract
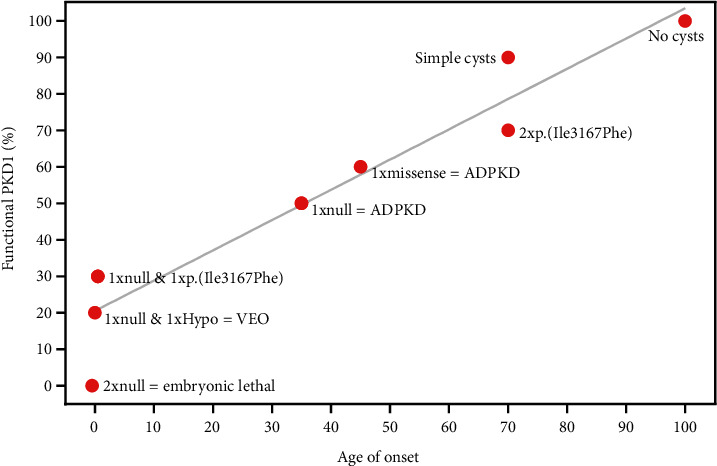
Biallelic PKD1 variants, including hypomorphic variants, can cause very early onset polycystic kidney disease (VEO-PKD). A family with unexplained recurrent VEO-PKD and neonatal demise in one dizygotic twin was referred for clinical testing. Further individuals with the putative hypomorphic PKD1 variant, p.(Ile3167Phe), were identified from the UK 100,000 genomes project (100 K), UK Biobank (UKBB), and a review of the literature. We identified a likely pathogenic PKD1 missense paternal variant and the putative hypomorphic PKD1 variant from the unaffected mother in the deceased twin but only the paternal PKD1 variant in the surviving dizygotic twin. Analysis of 100 K cases identified a second family with two siblings with similar biallelic inheritance who presented at birth with VEO-PKD and reached kidney failure in their teens unlike other affected relatives. Finally, a survey of 618 UKBB cases confirmed that adult patients monoallelic for PKD1 p.(Ile3167Phe) had normal kidney function. Our data reveals that p.(Ile3167Phe) is the second most common PKD1 hypomorphic variant identified and is neutral in heterozygosity but is associated with VEO-PKD when inherited in trans with a pathogenic PKD1 variant. Care should be taken to ensure that it is not automatically filtered from sequence data for VEO cases.
Copyright © 2023 Miranda Durkie et al.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures


References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
