

DIA-BERT: pre-trained end-to-end transformer models for enhanced DIA proteomics data analysis
- PMID: 40229248
- PMCID: PMC11997033
- DOI: 10.1038/s41467-025-58866-4
DIA-BERT: pre-trained end-to-end transformer models for enhanced DIA proteomics data analysis
Abstract
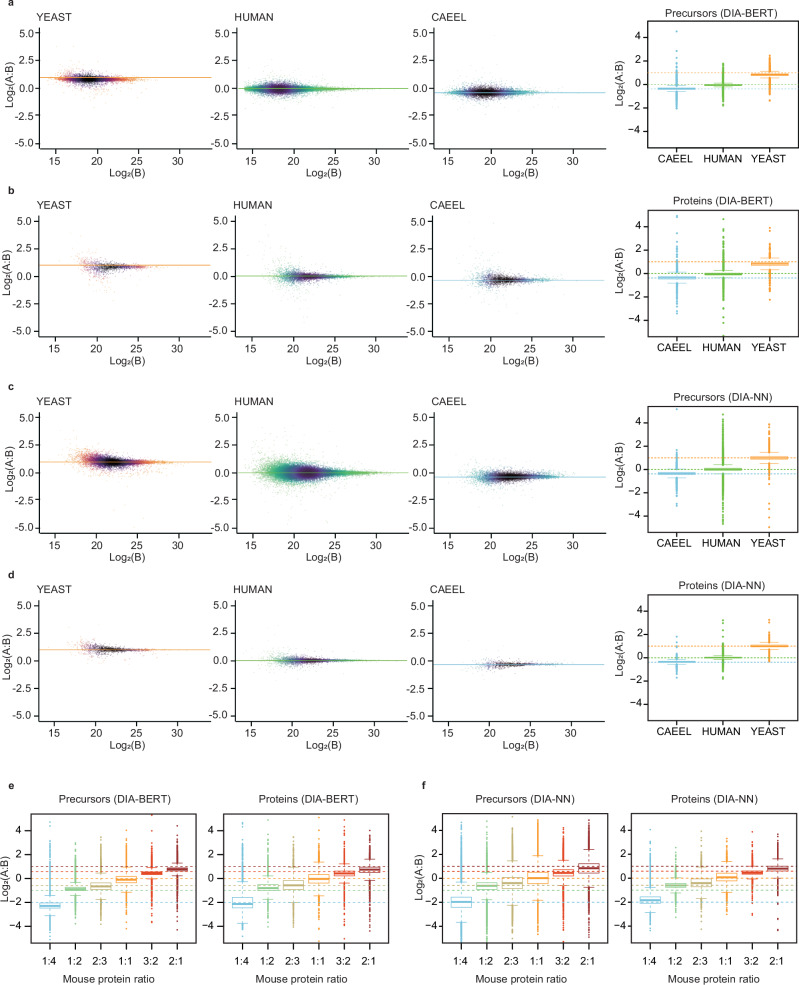
Data-independent acquisition mass spectrometry (DIA-MS) has become increasingly pivotal in quantitative proteomics. In this study, we present DIA-BERT, a software tool that harnesses a transformer-based pre-trained artificial intelligence (AI) model for analyzing DIA proteomics data. The identification model was trained using over 276 million high-quality peptide precursors extracted from existing DIA-MS files, while the quantification model was trained on 34 million peptide precursors from synthetic DIA-MS files. When compared to DIA-NN, DIA-BERT demonstrated a 51% increase in protein identifications and 22% more peptide precursors on average across five human cancer sample sets (cervical cancer, pancreatic adenocarcinoma, myosarcoma, gallbladder cancer, and gastric carcinoma), achieving high quantitative accuracy. This study underscores the potential of leveraging pre-trained models and synthetic datasets to enhance the analysis of DIA proteomics.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: T.G. is the founder of Westlake Omics (Hangzhou) Biotechnology Co., Ltd., while P.L. is staff of this company. The remaining authors declare no competing interests.
Figures

References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
