

Genomic epidemiology and phylodynamics of Acinetobacter baumannii bloodstream isolates in China
- PMID: 40229304
- PMCID: PMC11997098
- DOI: 10.1038/s41467-025-58772-9
Genomic epidemiology and phylodynamics of Acinetobacter baumannii bloodstream isolates in China
Abstract
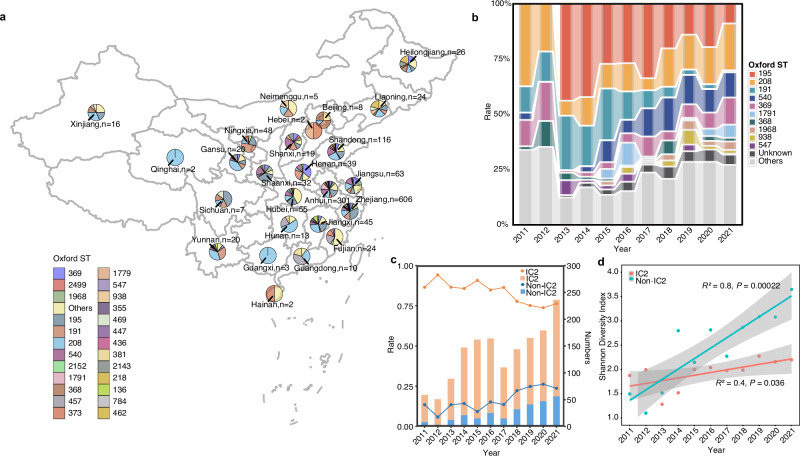
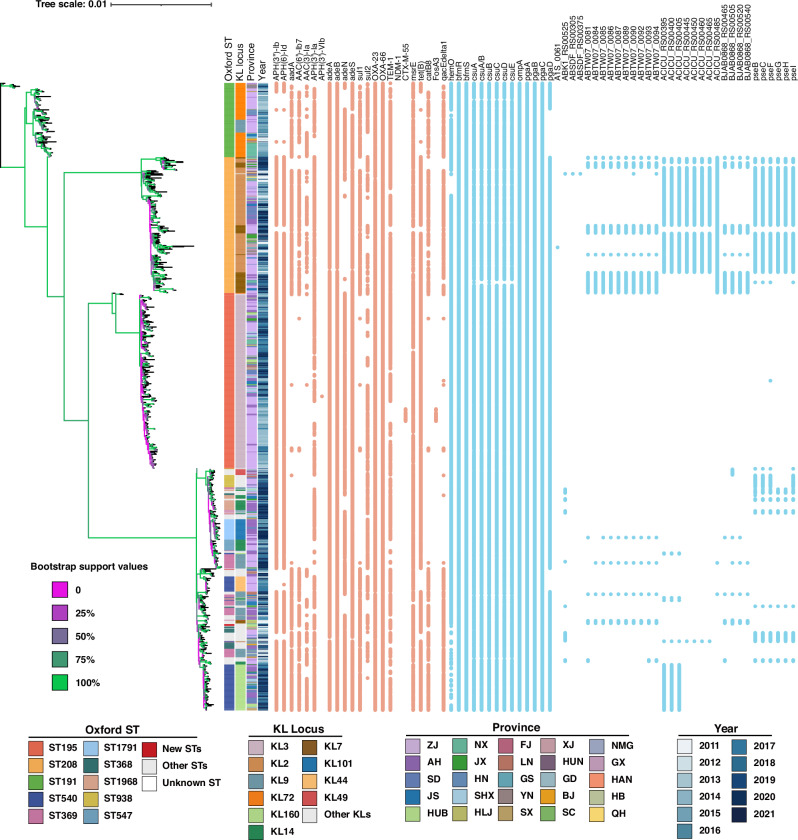
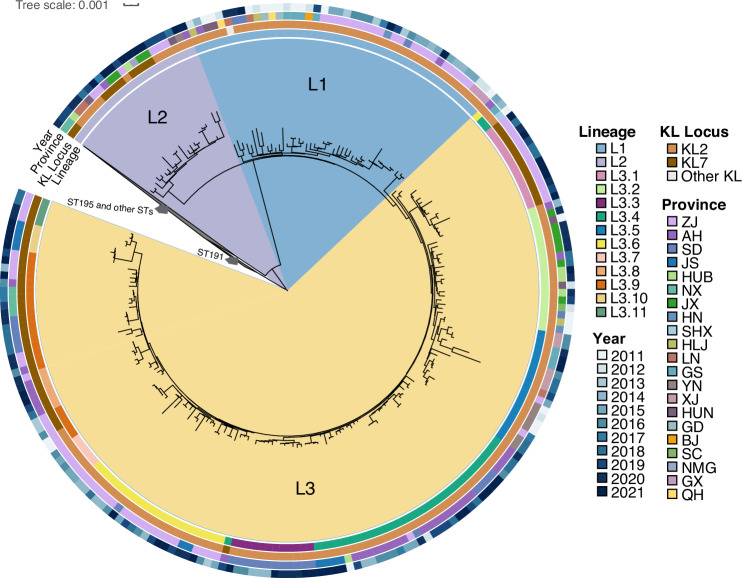
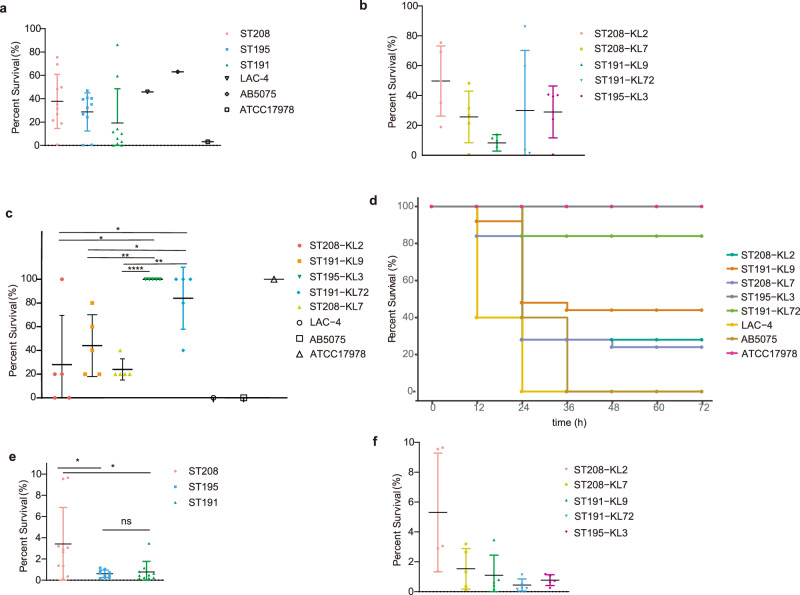
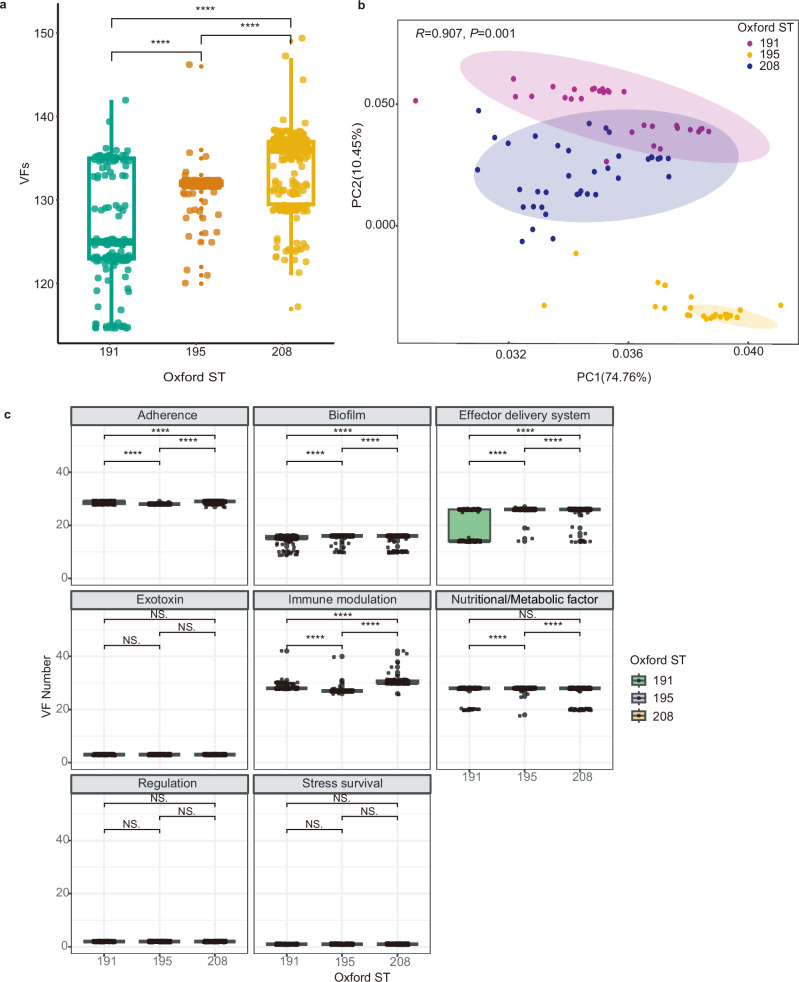
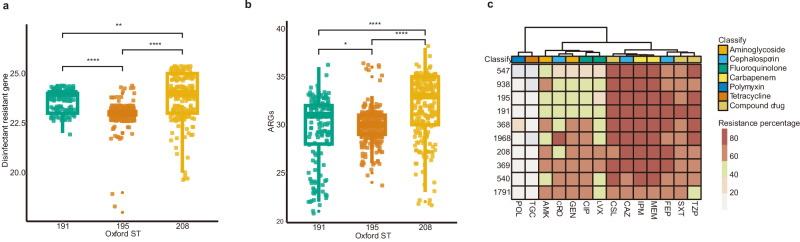
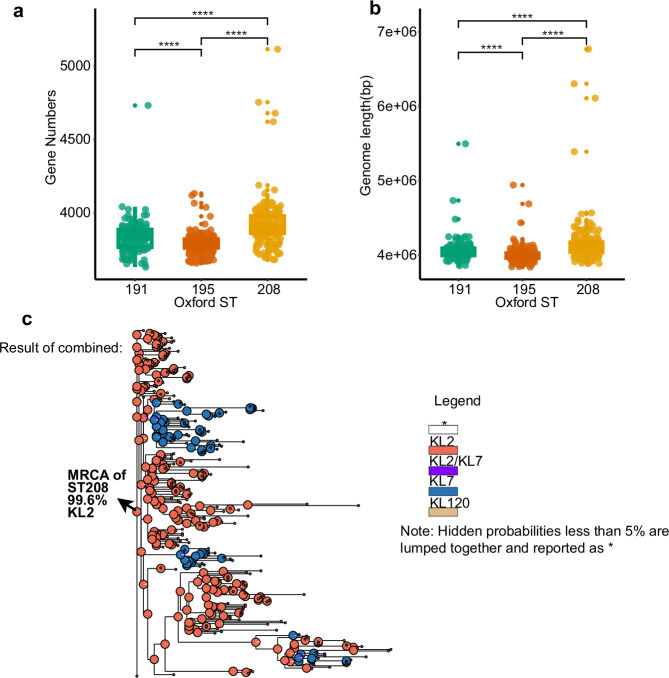
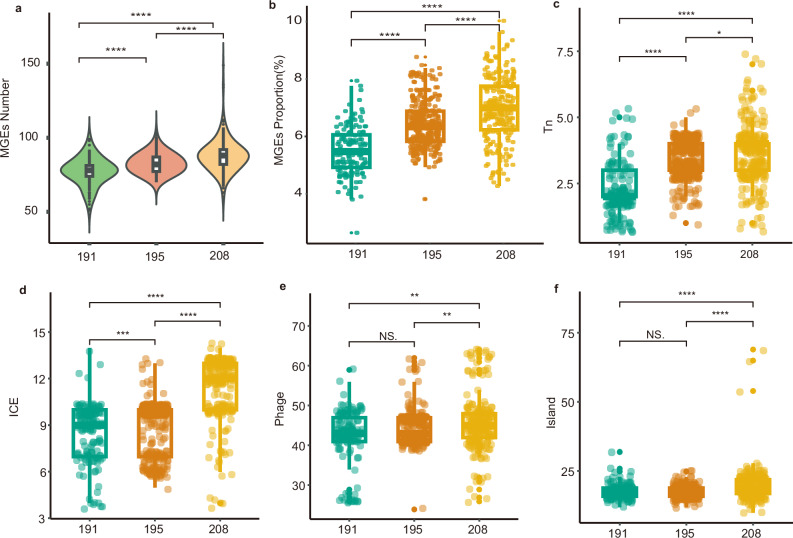
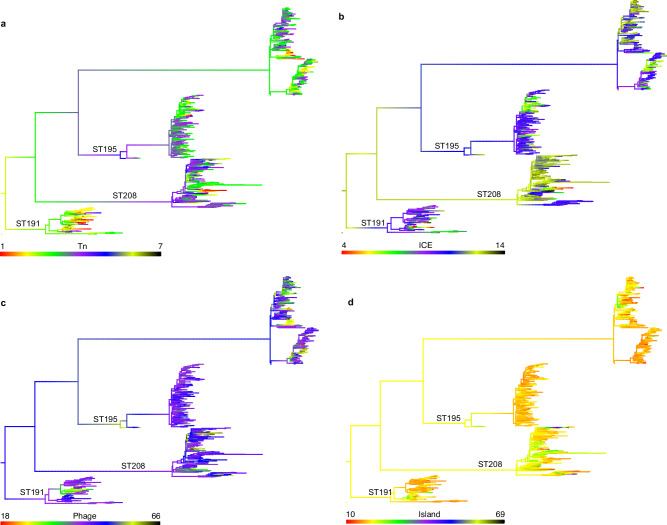
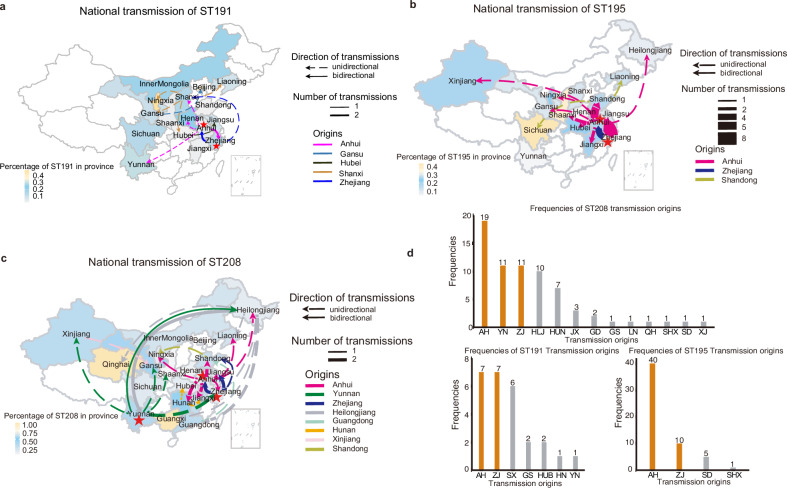
In recent decades, Acinetobacter baumannii has become a major global nosocomial pathogen, with bloodstream infections (BSIs) exhibiting mortality rates exceeding 60% and imposing substantial economic burdens. However, limited large-scale genomic epidemiology has hindered understanding of its population dynamics. Here, we analyzed 1506 non-repetitive BSI-causing A. baumannii isolates from 76 Chinese hospitals over a decade (2011-2021). We identified 149 sequence types (STs) and 101 K-locus types (KLs), revealing increased population diversity. International clone (IC) 2 accounted for 81.74% of isolates, with a notable shift in prevalent STs: ST208 increased while ST191 and ST195 declined, aligning with global trends. ST208 exhibited higher virulence, greater antibiotic resistance, enhanced desiccation tolerance, and more complex transmission patterns compared to ST191 and ST195. Its genomic plasticity drives its adaptation and spread. Using the high-resolution Oxford MLST scheme, this study uncovered greater diversity and genetic factors behind ST208's rise. A. baumannii is evolving from a low-virulence, multidrug-resistant pathogen to a more virulent one, highlighting the urgent need to address its growing threat. These findings have critical implications for infection control and public health policies.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
References
-
- Organization, W. H. Antimicrobial resistance surveillance in Europe 2022–2020 data. (World Health Organization. Regional Office for Europe, 2022).
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
