

A small-molecule inhibitor of BCL10-MALT1 interaction abrogates progression of diffuse large B cell lymphoma
- PMID: 40231473
- PMCID: PMC11996864
- DOI: 10.1172/JCI164573
A small-molecule inhibitor of BCL10-MALT1 interaction abrogates progression of diffuse large B cell lymphoma
Abstract
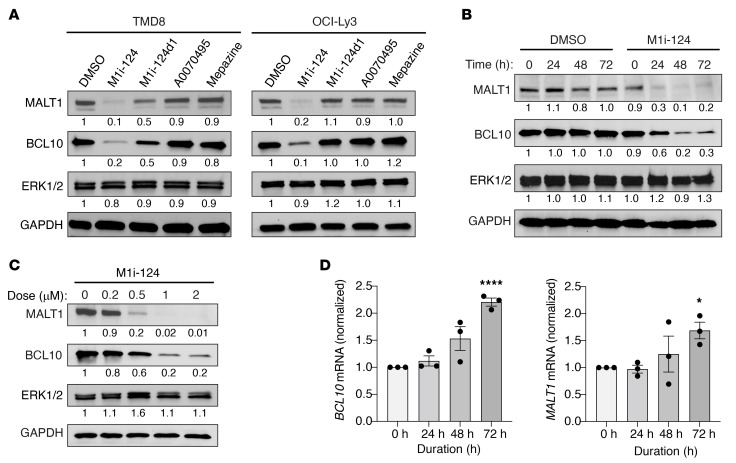
Diffuse large B cell lymphoma (DLBCL) is the most common type of non-Hodgkin lymphoma, and the activated B cell-like subtype (ABC-DLBCL) is associated with particularly poor outcome. Many ABC-DLBCLs harbor gain-of-function mutations that cause inappropriate assembly of the CARMA1-BCL10-MALT1 (CBM) signalosome, a cytoplasmic complex that drives downstream NF-κB signaling. MALT1 is the effector protein of the CBM signalosome such that its recruitment to the signalosome via interaction with BCL10 allows it to exert both protease and scaffolding activities that together synergize in driving NF-κB. Here, we demonstrate that a molecular groove located between two adjacent immunoglobulin-like domains within MALT1 represents a binding pocket for BCL10. Leveraging this discovery, we performed an in silico screen to identify small molecules that dock within this MALT1 groove and act as BCL10-MALT1 protein-protein interaction (PPI) inhibitors. We report the identification of M1i-124 as a first-in-class compound that blocks BCL10-MALT1 interaction, abrogates MALT1 scaffolding and protease activities, promotes degradation of BCL10 and MALT1 proteins, and specifically targets ABC-DLBCLs characterized by dysregulated MALT1. Our findings demonstrate that small-molecule inhibitors of BCL10-MALT1 interaction can function as potent agents to block MALT1 signaling in selected lymphomas, and provide a road map for clinical development of a new class of precision-medicine therapeutics.
Keywords: Immunology; Lymphomas; NF-kappaB; Oncology; Signal transduction; Therapeutics.
Figures








References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
