

Enhanced interpretation of immune cell phenotype and function through a rhesus macaque single-cell atlas
- PMID: 40233746
- PMCID: PMC12143338
- DOI: 10.1016/j.xgen.2025.100849
Enhanced interpretation of immune cell phenotype and function through a rhesus macaque single-cell atlas
Abstract
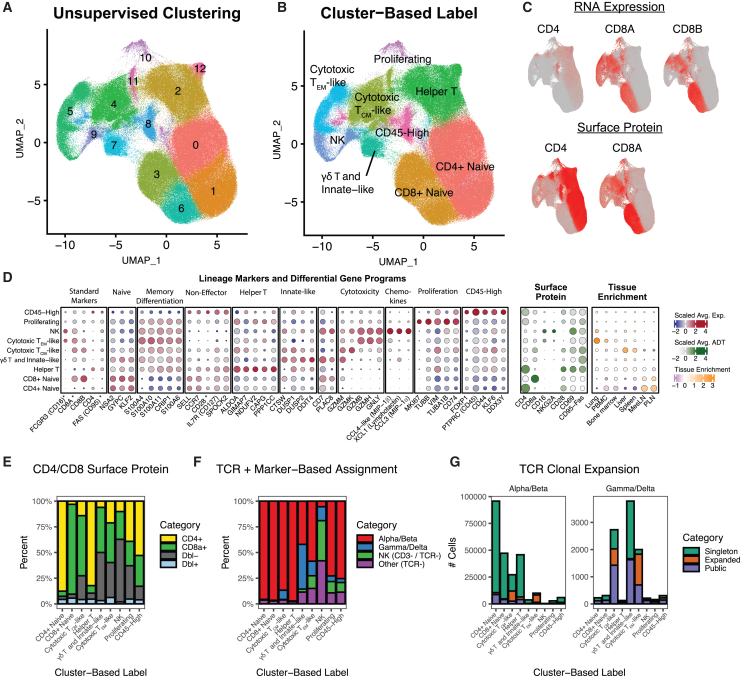
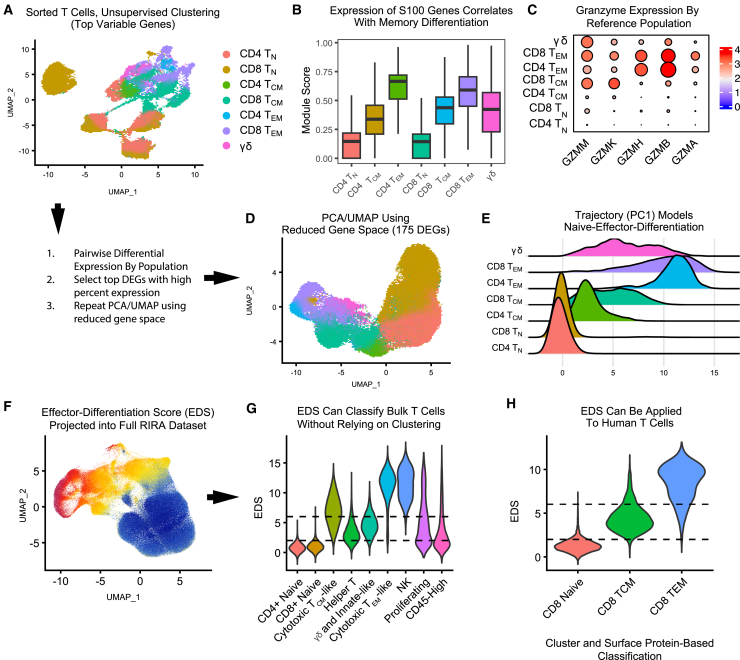
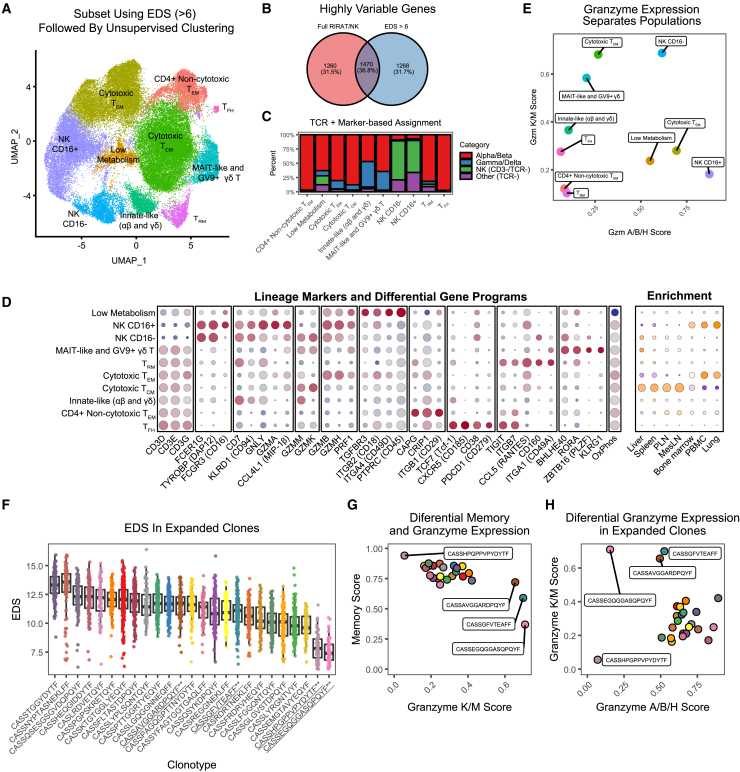
Single-cell RNA sequencing (scRNA-seq) allows cell classification using genome-wide transcriptional state; however, high-dimensional transcriptomic profiles, and the unsupervised analyses employed to interpret them, provide a systematically different view of biology than well-established functional/lineage definitions of immunocytes. Understanding these differences and limits is essential for accurate interpretation of these rich data. We present the Rhesus Immune Reference Atlas (RIRA), the first immune-focused macaque single-cell multi-tissue atlas. We contrasted transcriptional profiles against immune lineages, using surface protein and marker genes as ground truth. While the pattern of clustering can align with cell type, this is not always true. Especially within T and natural killer (NK) cells, many functionally distinct subsets lack defining markers, and strong shared expression programs, such as cytotoxicity, result in systematic intermingling by unsupervised clustering. We identified gene programs with high discriminatory/diagnostic value, including multi-gene signatures that model T/NK cell maturation. Directly measuring these diagnostic programs complements unsupervised analyses.
Keywords: T cells; immunology; rhesus macaque; single-cell RNA-seq.
Copyright © 2025 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
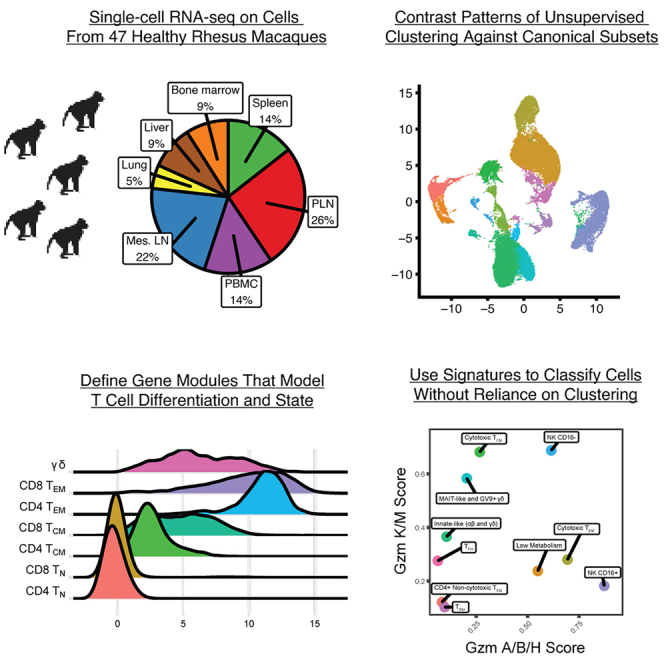
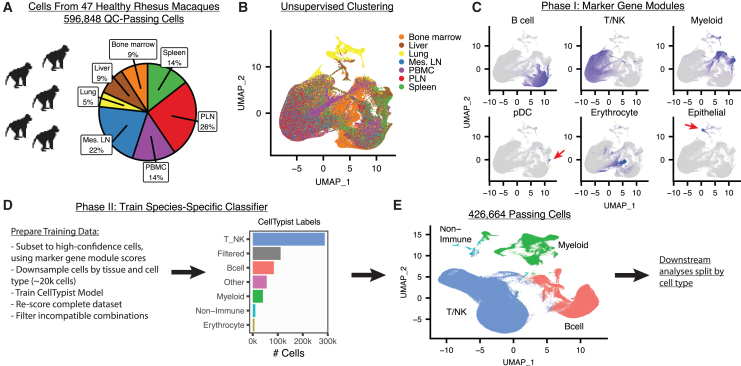
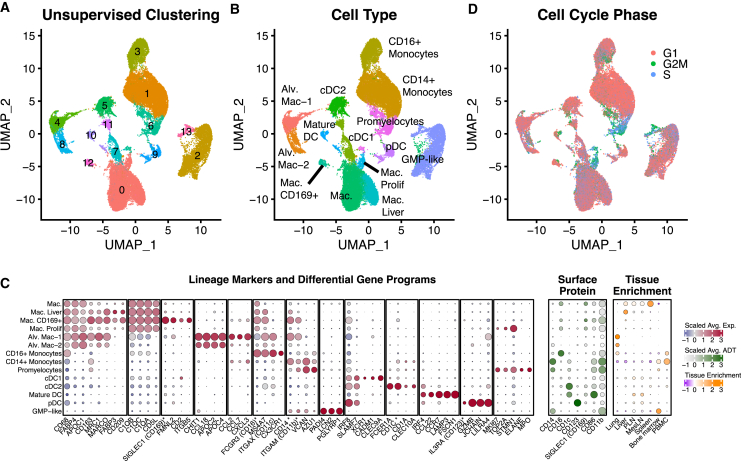
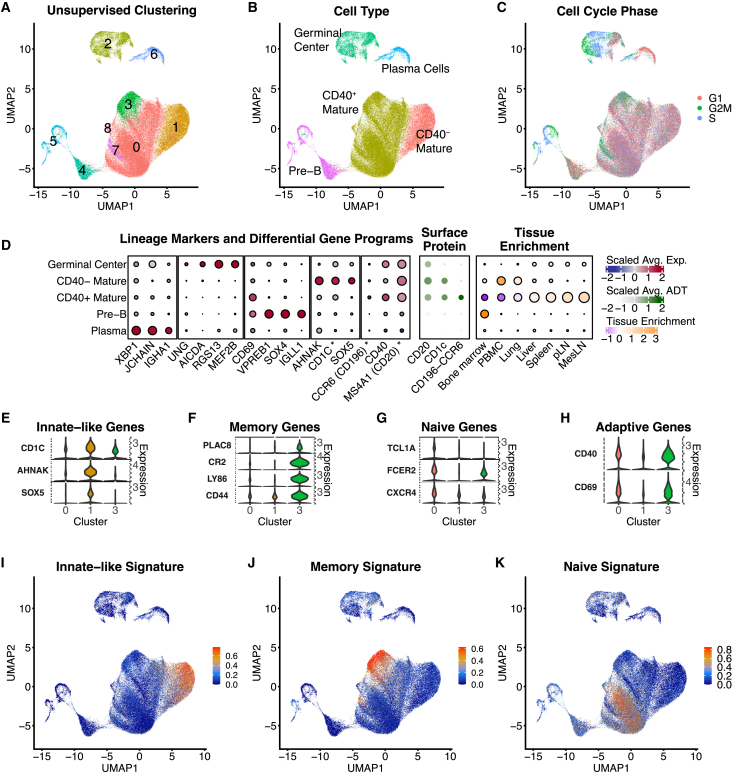
Similar articles
-
A scRNA-seq reference contrasting living and early post-mortem human retina across diverse donor states.Hum Genomics. 2025 Jul 14;19(1):81. doi: 10.1186/s40246-025-00796-9. Hum Genomics. 2025. PMID: 40660409 Free PMC article.
-
New insights for precision treatment of glioblastoma from analysis of single-cell lncRNA expression.J Cancer Res Clin Oncol. 2021 Jul;147(7):1881-1895. doi: 10.1007/s00432-021-03584-9. Epub 2021 Mar 11. J Cancer Res Clin Oncol. 2021. PMID: 33693962 Free PMC article.
-
Reference Vector-guided Evolutionary Algorithm for cluster analysis of single-cell transcriptomes.Comput Methods Programs Biomed. 2025 Sep;269:108873. doi: 10.1016/j.cmpb.2025.108873. Epub 2025 Jun 6. Comput Methods Programs Biomed. 2025. PMID: 40499345
-
The Lived Experience of Autistic Adults in Employment: A Systematic Search and Synthesis.Autism Adulthood. 2024 Dec 2;6(4):495-509. doi: 10.1089/aut.2022.0114. eCollection 2024 Dec. Autism Adulthood. 2024. PMID: 40018061 Review.
-
Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19.Cochrane Database Syst Rev. 2022 May 20;5(5):CD013665. doi: 10.1002/14651858.CD013665.pub3. Cochrane Database Syst Rev. 2022. PMID: 35593186 Free PMC article.
References
-
- Murphy K., Weaver C. 9th edition. Garland Science/Taylor & Francis Group, LLC; 2016. Janeway's Immunobiology.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
