

Allele age estimators designed for whole-genome datasets show only a moderate reduction in performance when applied to whole-exome datasets
- PMID: 40238934
- PMCID: PMC12135005
- DOI: 10.1093/g3journal/jkaf088
Allele age estimators designed for whole-genome datasets show only a moderate reduction in performance when applied to whole-exome datasets
Abstract
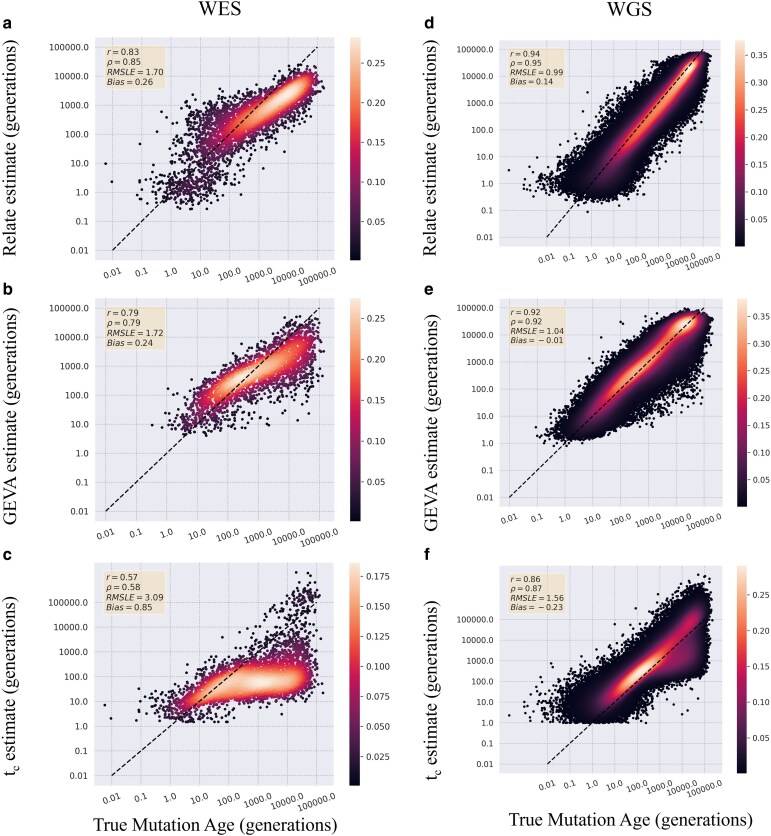
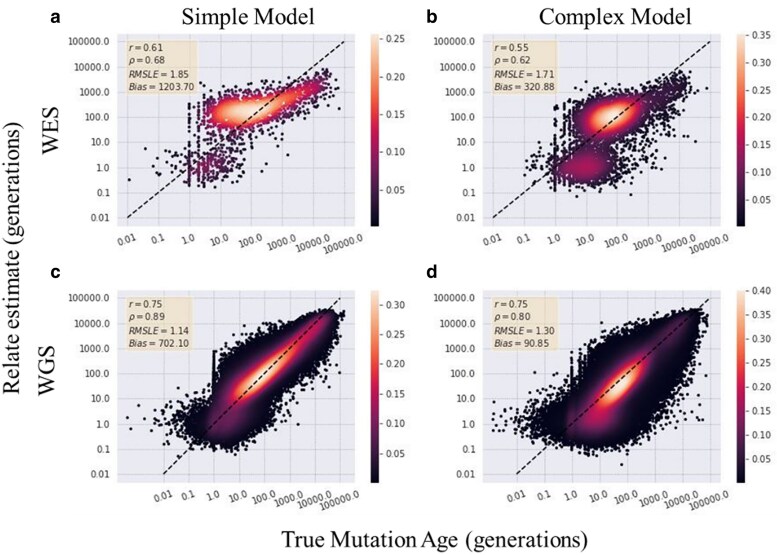
As personalized genomics becomes more affordable, larger numbers of rare variants are being discovered, leading to important initiatives in identifying the functional impacts in relation to disease phenotypes. One way to characterize these variants is to estimate the time the mutation entered the population. However, allele age estimators such as those implemented in the programs Relate, Genealogical Estimator of Variant Age, and Runtc were developed based on the assumption that datasets include the entire genome. We examined the performance of each of these estimators on simulated exome data under a neutral constant population size model, as well as under population expansion and background selection models. We found that each provides usable estimates of allele age from whole-exome datasets. Relate performs the best amongst all 3 estimators with Pearson coefficients of 0.83 and 0.73 (with respect to true simulated values for neutral constant and expansion population models, respectively) with a 12% and 20% decrease in correlation between whole-genome and whole-exome estimations. Of the 3 estimators, Relate is best able to parallelize to yield quick results with little resources; however, Relate is currently only able to scale to thousands of samples making it unable to match the hundreds of thousands of samples being currently released. While more work is needed to expand the capabilities of current methods of estimating allele age, these methods show a modest decrease in performance in the estimation of the age of mutations.
Keywords: WES; allele age; exome; personalized genomics; whole-exome sequencing.
© The Author(s) 2025. Published by Oxford University Press on behalf of The Genetics Society of America.
Conflict of interest statement
Conflicts of interest: The author(s) declare no conflict of interest.
Figures



Update of
-
Allele age estimators designed for whole genome datasets show only a moderate reduction in performance when applied to whole exome datasets.bioRxiv [Preprint]. 2025 Mar 3:2024.02.01.578465. doi: 10.1101/2024.02.01.578465. bioRxiv. 2025. Update in: G3 (Bethesda). 2025 Jun 4;15(6):jkaf088. doi: 10.1093/g3journal/jkaf088. PMID: 38370640 Free PMC article. Updated. Preprint.
Similar articles
-
Allele age estimators designed for whole genome datasets show only a moderate reduction in performance when applied to whole exome datasets.bioRxiv [Preprint]. 2025 Mar 3:2024.02.01.578465. doi: 10.1101/2024.02.01.578465. bioRxiv. 2025. Update in: G3 (Bethesda). 2025 Jun 4;15(6):jkaf088. doi: 10.1093/g3journal/jkaf088. PMID: 38370640 Free PMC article. Updated. Preprint.
-
JWES: a new pipeline for whole genome/exome sequence data processing, management, and gene-variant discovery, annotation, prediction, and genotyping.FEBS Open Bio. 2021 Sep;11(9):2441-2452. doi: 10.1002/2211-5463.13261. Epub 2021 Aug 11. FEBS Open Bio. 2021. PMID: 34370400 Free PMC article.
-
Benchmarking of variant calling software for whole-exome sequencing using gold standard datasets.Sci Rep. 2025 Apr 21;15(1):13697. doi: 10.1038/s41598-025-97047-7. Sci Rep. 2025. PMID: 40258889 Free PMC article.
-
Frequency and management of medically actionable incidental findings from genome and exome sequencing data: a systematic review.Physiol Genomics. 2021 Sep 1;53(9):373-384. doi: 10.1152/physiolgenomics.00025.2021. Epub 2021 Jul 12. Physiol Genomics. 2021. PMID: 34250816
-
Added Value of Reanalysis of Whole Exome- and Whole Genome Sequencing Data From Patients Suspected of Primary Immune Deficiency Using an Extended Gene Panel and Structural Variation Calling.Front Immunol. 2022 Jun 30;13:906328. doi: 10.3389/fimmu.2022.906328. eCollection 2022. Front Immunol. 2022. PMID: 35874679 Free PMC article. Review.
References
-
- Almogy G, Pratt M, Oberstrass F, Lee L, Mazur D, Beckett N, Barad O, Soifer I, Perelman E, Etzioni Y, et al. 2022. Cost-efficient whole genome-sequencing using novel mostly natural sequencing-by-synthesis chemistry and open fluidics platform. BioRvix 2022.05.29.493900. doi:10.1101/2022.05.29.493900. https://www.biorxiv.org/content/10.1101/2022.05.29.493900v4. - DOI - DOI
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials