

Herpes simplex virus 1 encodes a STING antagonist that can be therapeutically targeted
- PMID: 40239620
- PMCID: PMC12047521
- DOI: 10.1016/j.xcrm.2025.102051
Herpes simplex virus 1 encodes a STING antagonist that can be therapeutically targeted
Abstract
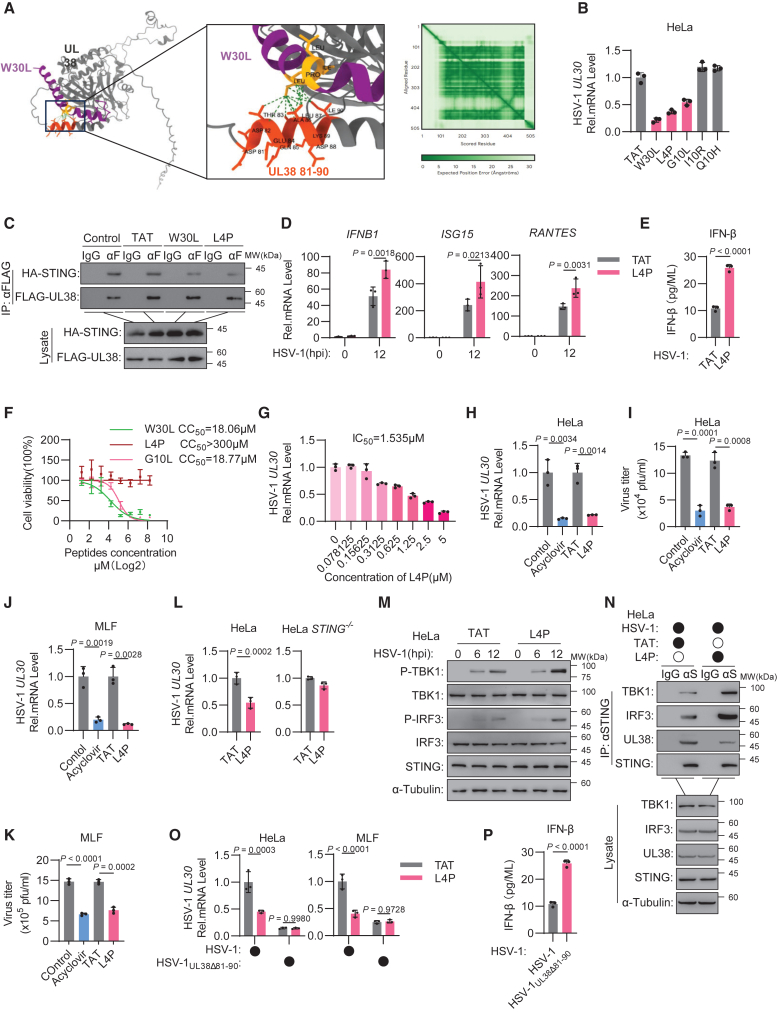
Herpes simplex virus 1 (HSV-1) is a ubiquitous human pathogen that causes serious symptoms and is known for its strong interactions with host immunity. Here, we revealed that the HSV-1-encoded UL38 is a stimulator of interferon genes (STING) antagonist that interacts with STING to abrogate the STING-TANK-binding kinase 1 (TBK1)-interferon regulatory factor 3 (IRF3) interaction, thereby suppressing cyclic GMP-AMP synthase (cGAS)-STING-dependent immune signaling. Losing UL38's STING antagonist activity made HSV-1 incapable of immune evasion and less replicable and pathogenic in vivo. Moreover, on the basis of the UL38-interacting sequence within STING, we rationally designed a series of peptides to target the STING-UL38 interface of UL38 specifically. Among them, a peptide effectively disrupts the STING-UL38 interaction, which unlocks the UL38-suppressed immune response and shows potent therapeutic efficacy against HSV-1 infection in vivo. Therefore, our findings demonstrate that HSV-1 UL38 is a STING antagonist, and targeting the activity of UL38 is a promising strategy for the development of antivirals against this notorious virus.
Keywords: STING; UL38; antiviral innate immunity; antivirals; drug target; herpes simplex virus 1.
Copyright © 2025 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests Wuhan Institute of Virology on behalf of Y.R., X.Z., and A.W. has filed a patent application for the use of antiviral peptide (Chinese patent application 202510237389.X).
Figures







References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
