

Systematic genetic perturbation reveals principles underpinning robustness of the epigenetic regulatory network
- PMID: 40239999
- PMCID: PMC12000879
- DOI: 10.1093/nar/gkaf297
Systematic genetic perturbation reveals principles underpinning robustness of the epigenetic regulatory network
Abstract
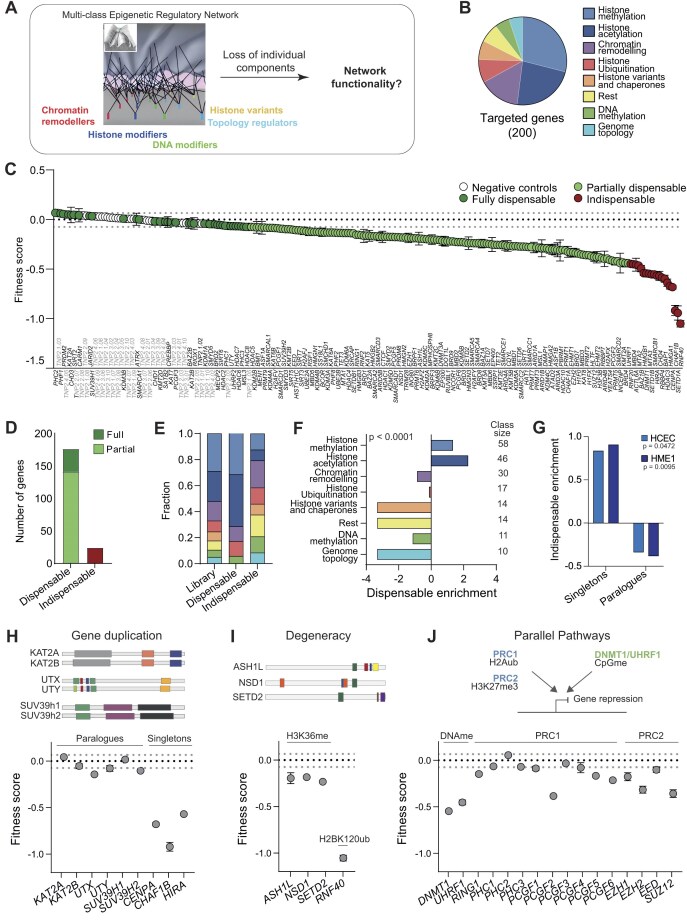
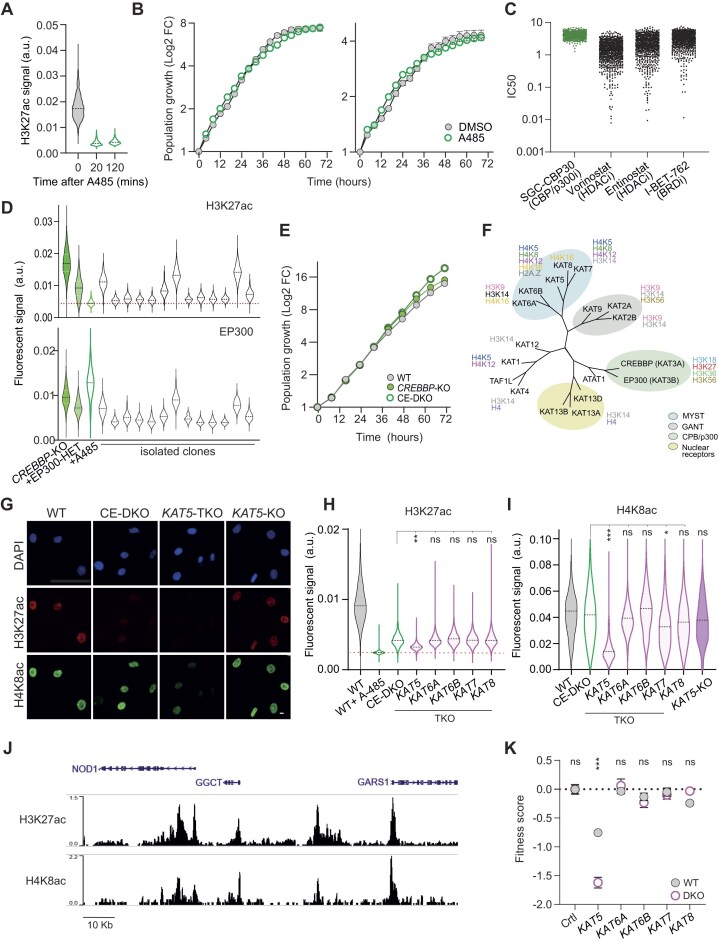
The molecular control of epigenetic information relies on hundreds of proteins of diverse function, which cooperate in defining chromatin structure and DNA methylation landscapes. While many individual pathways have been characterized, how different classes of epigenetic regulators interact to build a resilient epigenetic regulatory network (ERN) remains poorly understood. Here, we show that most individual regulators are dispensable for somatic cell fitness, and that robustness emerges from multiple layers of functional cooperation and degeneracy among network components. By disrupting 200 epigenetic regulator genes, individually or in combination, we generated network-wide maps of functional interactions for representative regulators. We found that paralogues represent only a first layer of functional compensation within the ERN, with intra- or inter-class interactions buffering the effects of perturbation in a gene-specific manner: while CREBBP cooperates with multiple acetyltransferases to form a subnetwork that ensures robust chromatin acetylation, ARID1A interacts with regulators from across all functional classes. When combined with oncogene activation, the accumulated epigenetic disorder exposes a synthetic fragility and broadly sensitizes ARID1A-deficient cells to further perturbation. Our findings reveal homeostatic mechanisms through which the ERN sustains somatic cell fitness and uncover how the network remodels as the epigenome is progressively deregulated in disease.
Plain language summary
Cells regulate their DNA through a network of proteins that shape chromatin structure and modify chemical markers. While many pathways have been studied individually, how they interact to maintain stability remains unclear. We investigated this by disrupting 200 genes involved in epigenetic regulation, one by one or in combination, to reveal their interactions. We found that most regulators are nonessential due to a variety of backup mechanisms. However, cancer-related mutations weaken this network, making cells more vulnerable to further disruption. Overall, our work helps explain how cells maintain stability despite disruptions and how this balance shifts in disease, potentially revealing new opportunities for treatment.
© The Author(s) 2025. Published by Oxford University Press on behalf of Nucleic Acids Research.
Conflict of interest statement
None declared.
Figures




Similar articles
-
'Traffic light rules': Chromatin states direct miRNA-mediated network motifs running by integrating epigenome and regulatome.Biochim Biophys Acta. 2016 Jul;1860(7):1475-88. doi: 10.1016/j.bbagen.2016.04.008. Epub 2016 Apr 14. Biochim Biophys Acta. 2016. PMID: 27091612
-
Identification of a transcriptional signature found in multiple models of ASD and related disorders.Genome Res. 2022 Sep 27;32(9):1642-1654. doi: 10.1101/gr.276591.122. Genome Res. 2022. PMID: 36104286 Free PMC article.
-
Compromised epigenetic robustness in cancer: fueling evolution, exposing weakness.Trends Cancer. 2025 Jun;11(6):575-590. doi: 10.1016/j.trecan.2025.02.001. Epub 2025 Mar 6. Trends Cancer. 2025. PMID: 40055120 Review.
-
Epigenetic regulation of stem cells : the role of chromatin in cell differentiation.Adv Exp Med Biol. 2013;786:307-28. doi: 10.1007/978-94-007-6621-1_17. Adv Exp Med Biol. 2013. PMID: 23696364
-
ARID1A mutations in cancer: another epigenetic tumor suppressor?Cancer Discov. 2013 Jan;3(1):35-43. doi: 10.1158/2159-8290.CD-12-0361. Epub 2012 Dec 3. Cancer Discov. 2013. PMID: 23208470 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
