

Histone chaperones coupled to DNA replication and transcription control divergent chromatin elements to maintain cell fate
- PMID: 40240143
- PMCID: PMC12047658
- DOI: 10.1101/gad.352316.124
Histone chaperones coupled to DNA replication and transcription control divergent chromatin elements to maintain cell fate
Abstract
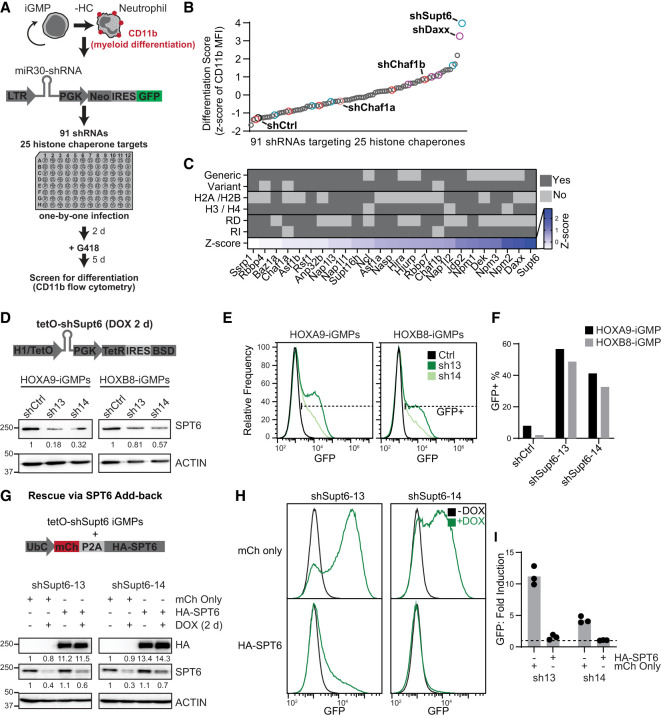
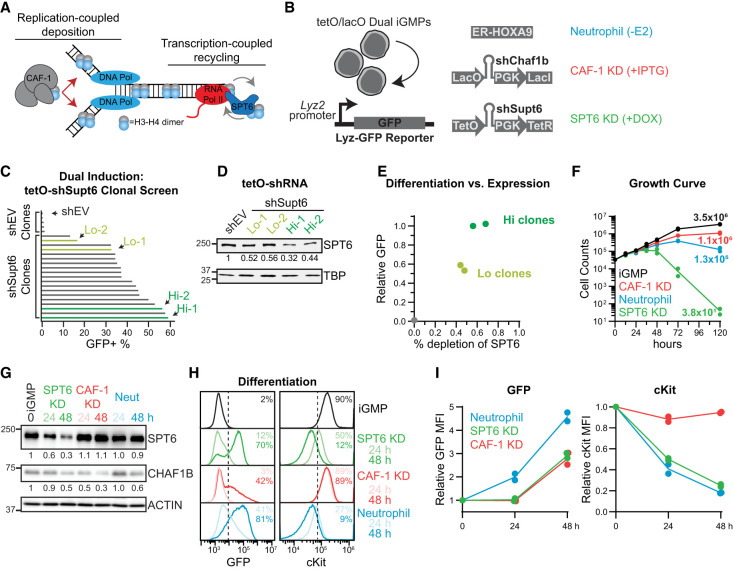
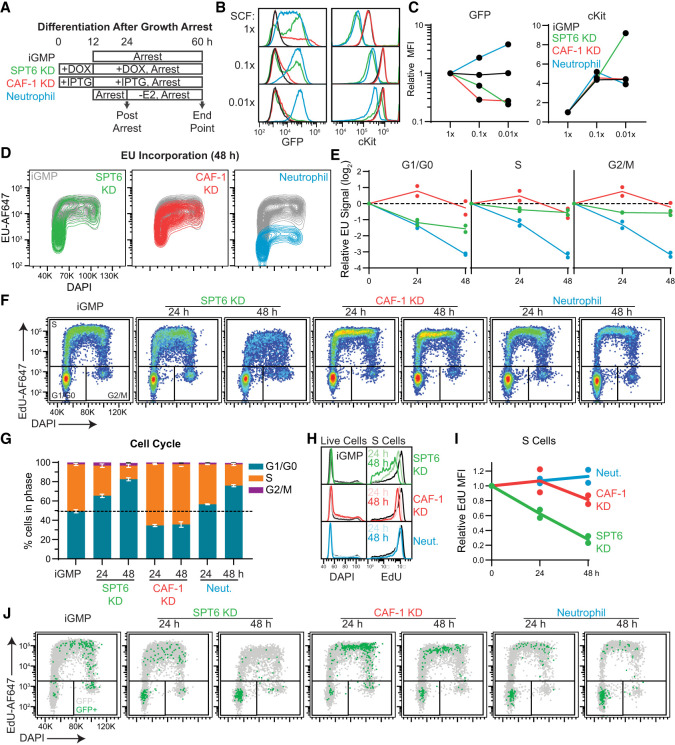
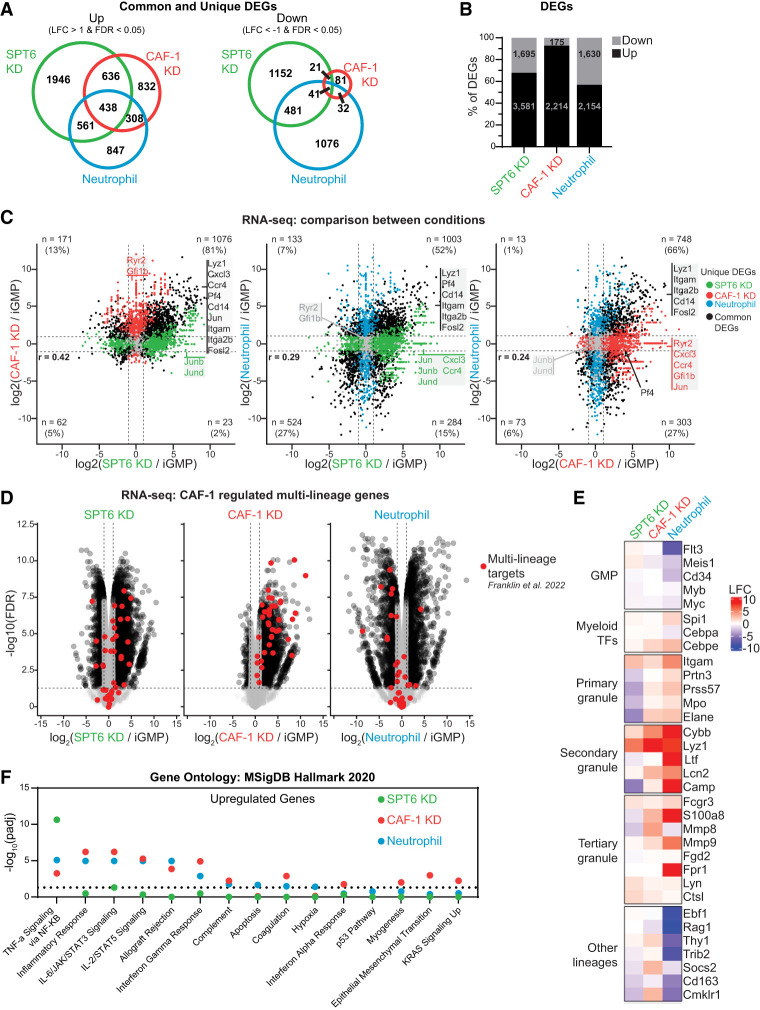
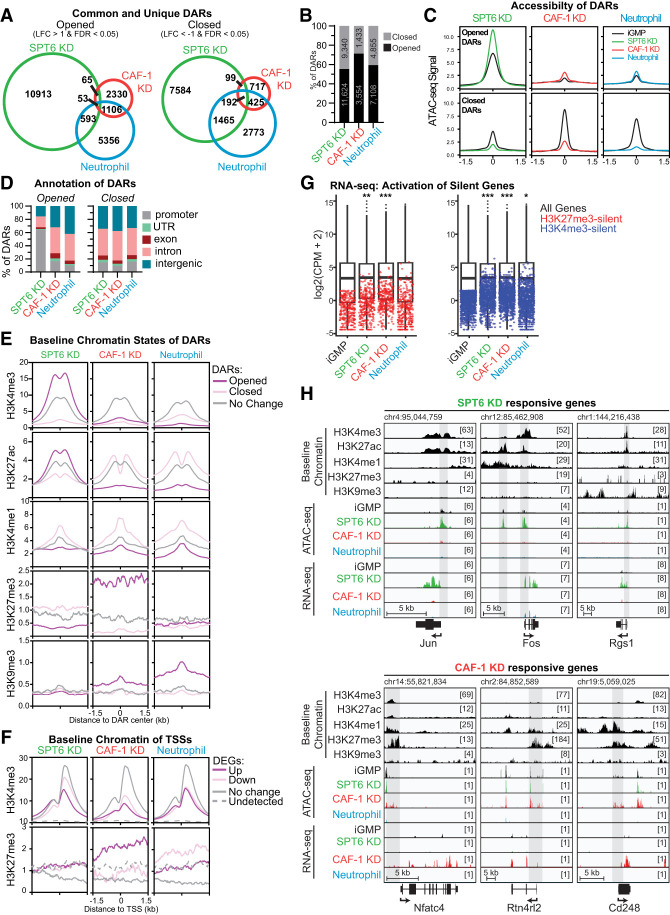
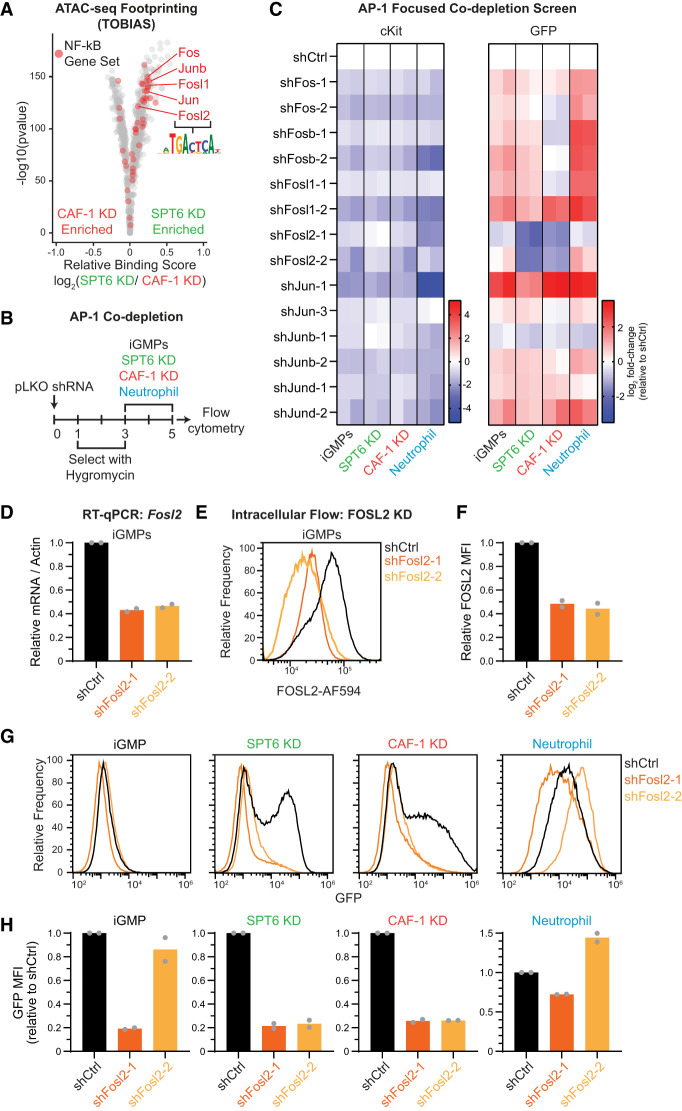
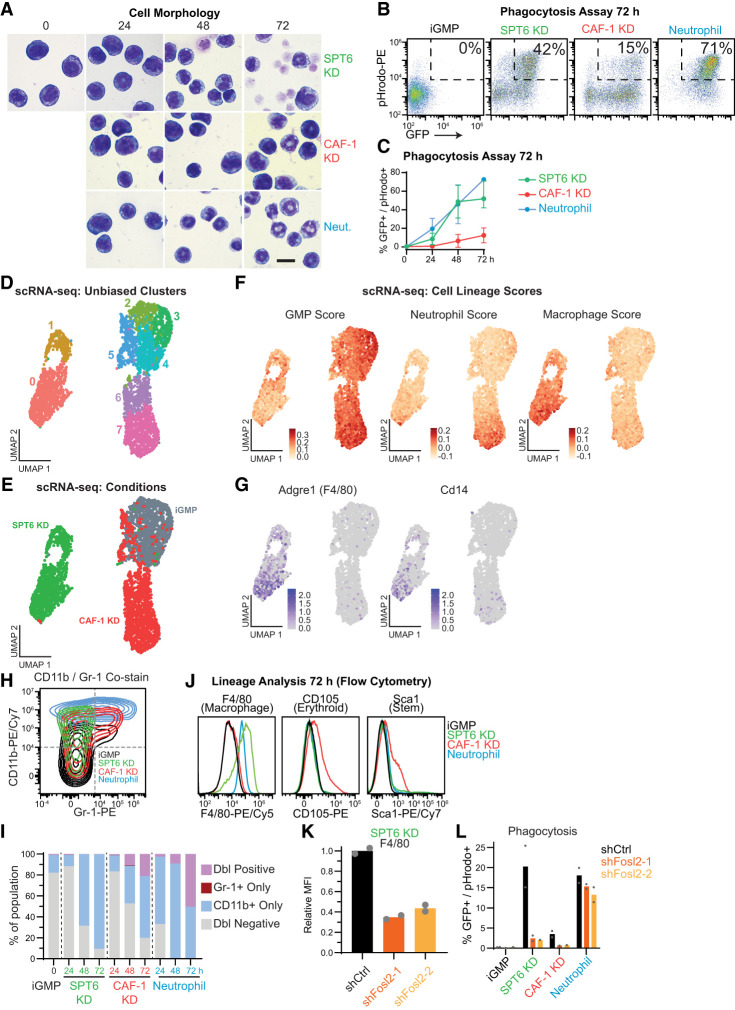
The manipulation of DNA replication and transcription can be harnessed to control cell fate. Central to the regulation of these DNA-templated processes are histone chaperones, which in turn are emerging as cell fate regulators. Histone chaperones are a group of proteins with diverse functions that are primarily involved in escorting histones to assemble nucleosomes and maintain the chromatin landscape. Whether distinct histone chaperone pathways control cell fate and whether they function using related mechanisms remain unclear. To address this, we performed a screen to assess the requirement of diverse histone chaperones in the self-renewal of hematopoietic stem and progenitor cells. Remarkably, all candidates were required to maintain cell fate to differing extents, with no clear correlation with their specific histone partners or DNA-templated process. Among all the histone chaperones, the loss of the transcription-coupled histone chaperone SPT6 most strongly promoted differentiation, even more than the major replication-coupled chromatin assembly factor complex CAF-1. To directly compare how DNA replication- and transcription-coupled histone chaperones maintain stem cell self-renewal, we generated an isogenic dual-inducible system to perturb each pathway individually. We found that SPT6 and CAF-1 perturbations required cell division to induce differentiation but had distinct effects on cell cycle progression, chromatin accessibility, and lineage choice. CAF-1 depletion led to S-phase accumulation, increased heterochromatic accessibility (particularly at H3K27me3 sites), and aberrant multilineage gene expression. In contrast, SPT6 loss triggered cell cycle arrest, altered accessibility at promoter elements, and drove lineage-specific differentiation, which is in part influenced by AP-1 transcription factors. Thus, CAF-1 and SPT6 histone chaperones maintain cell fate through distinct mechanisms, highlighting how different chromatin assembly pathways can be leveraged to alter cell fate.
Keywords: DNA replication; histone chaperones; lineage choice; stem cells; transcription; transcription factors.
© 2025 Franklin et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Abe Y, Kofman ER, Ouyang Z, Cruz-Becerra G, Spann NJ, Seidman JS, Troutman TD, Stender JD, Taylor H, Fan W, et al. 2024. A TLR4/TRAF6-dependent signaling pathway mediates NCoR coactivator complex formation for inflammatory gene activation. Proc Natl Acad Sci 121: e2316104121. 10.1073/pnas.2316104121 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
- F30 HL156404/HL/NHLBI NIH HHS/United States
- R35 GM151016/GM/NIGMS NIH HHS/United States
- S10 OD021718/OD/NIH HHS/United States
- P30 CA045508/CA/NCI NIH HHS/United States
- S10 OD010794/OD/NIH HHS/United States
- R00 HD099316/HD/NICHD NIH HHS/United States
- P30 CA062203/CA/NCI NIH HHS/United States
- R01 GM144693/GM/NIGMS NIH HHS/United States
- R01 CA281106/CA/NCI NIH HHS/United States
- S10 RR025496/RR/NCRR NIH HHS/United States
- R01 CA229699/CA/NCI NIH HHS/United States
- R35 CA242379/CA/NCI NIH HHS/United States
- T32 GM144273/GM/NIGMS NIH HHS/United States
- R35 GM151004/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources