

Nanobody Engineered and Photosensitiser Loaded Bacterial Outer Membrane Vesicles Potentiate Antitumour Immunity and Immunotherapy
- PMID: 40240911
- PMCID: PMC12003094
- DOI: 10.1002/jev2.70069
Nanobody Engineered and Photosensitiser Loaded Bacterial Outer Membrane Vesicles Potentiate Antitumour Immunity and Immunotherapy
Abstract
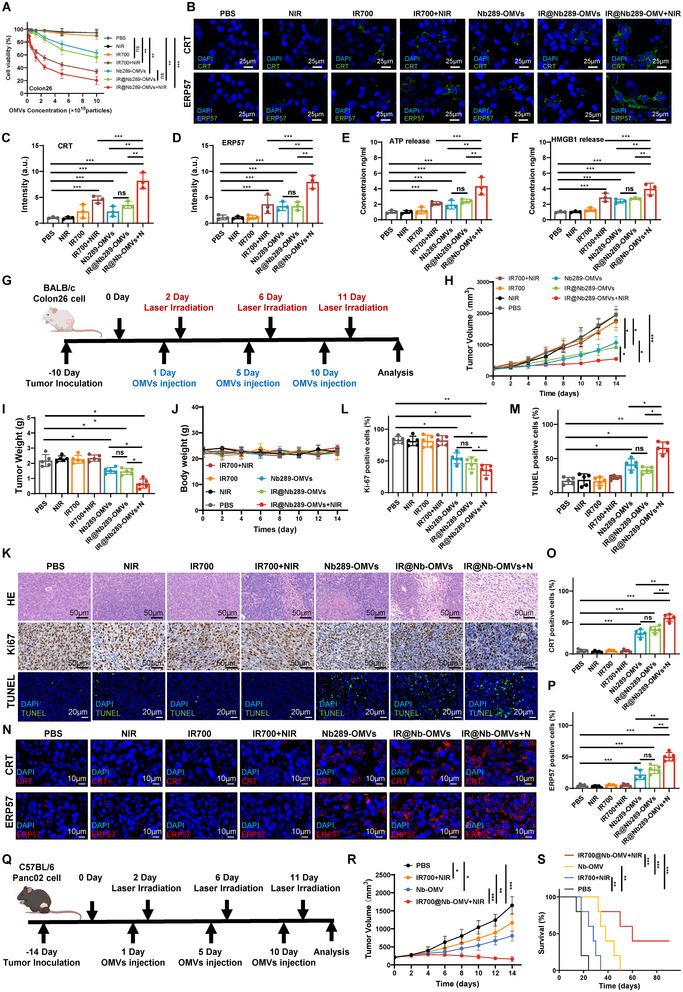
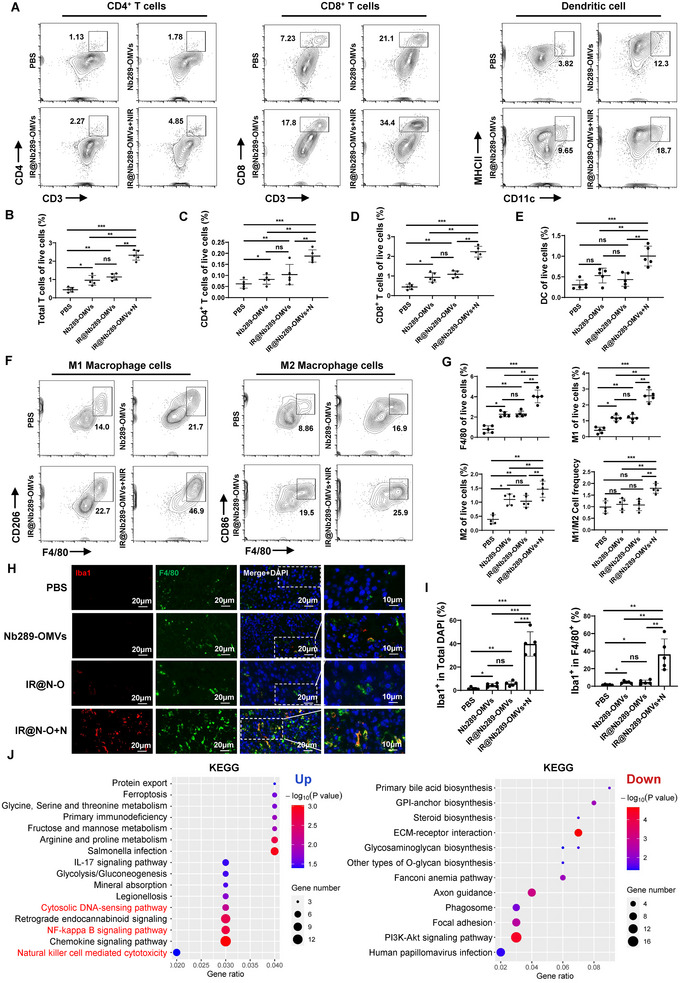
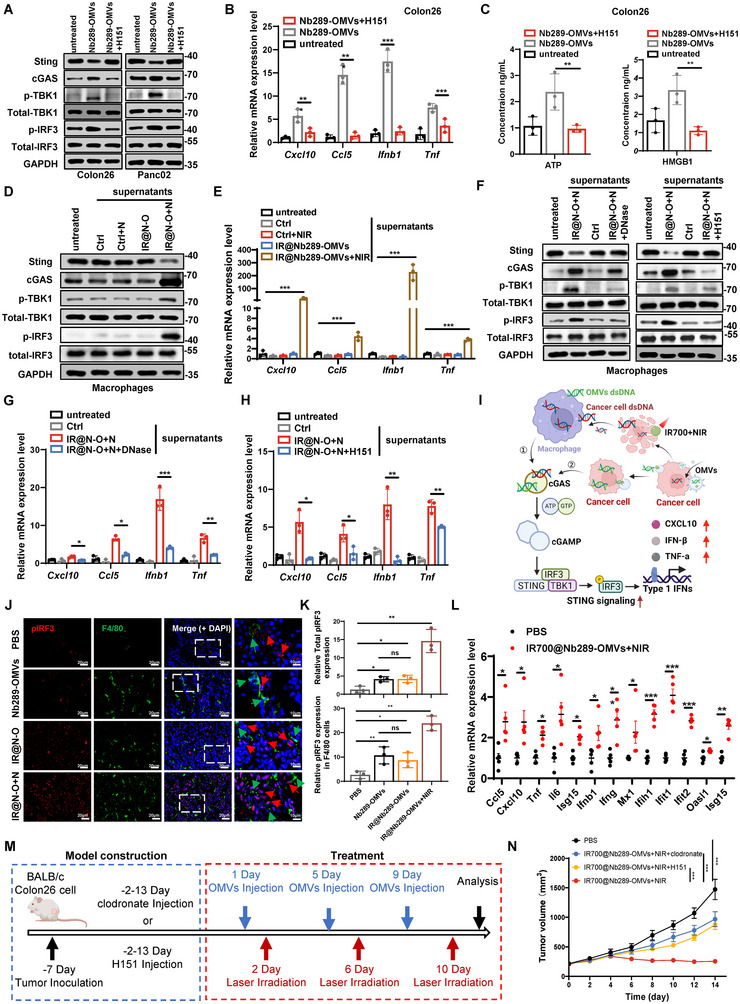
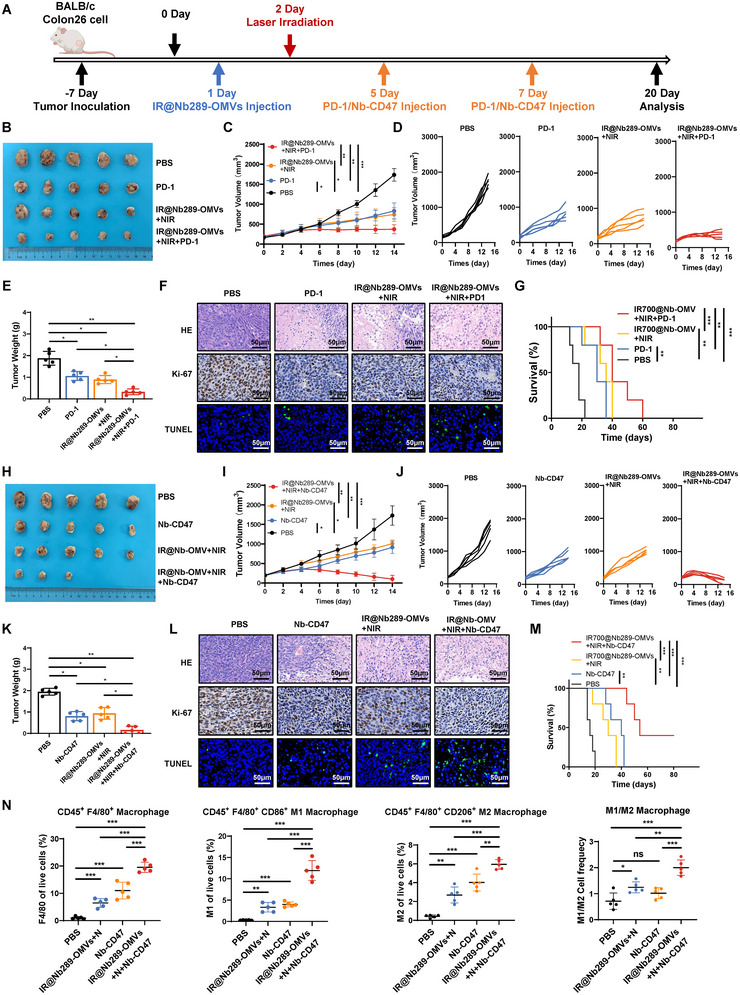
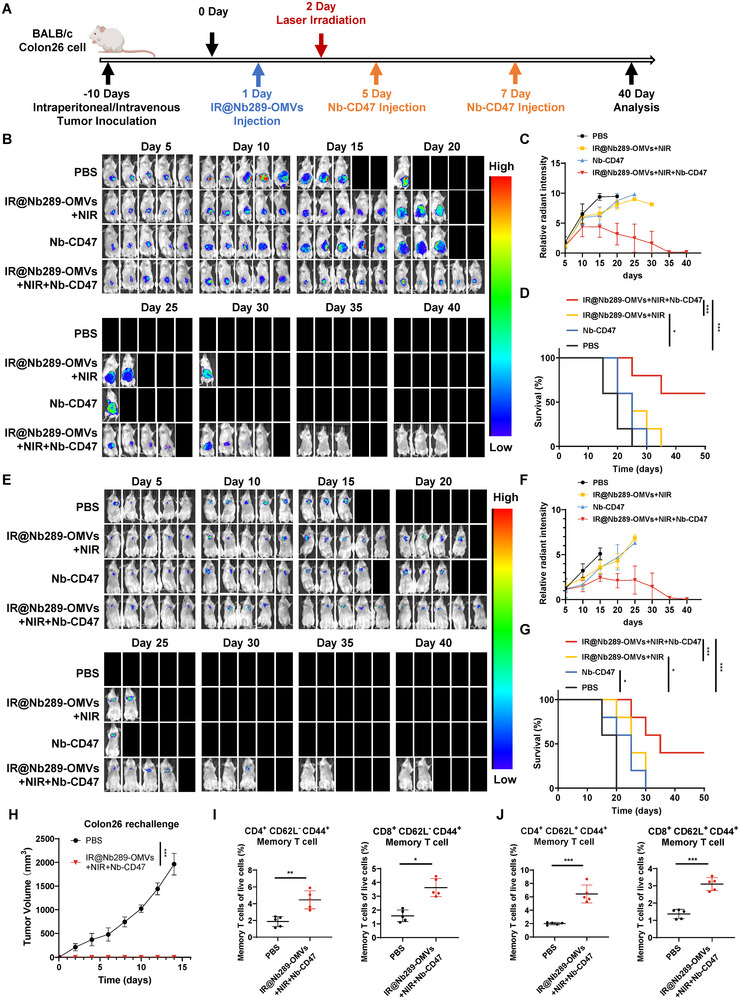
Bacterial outer membrane vesicles (OMVs) are promising as antitumour agents, but their clinical application is limited by toxicity concerns and unclear mechanisms. We engineered OMVs with cadherin 17 (CDH17) tumour-targeting nanobodies, enhancing tumour selectivity and efficacy while reducing adverse effects. These engineered OMVs function as natural stimulator of interferon genes (STING) agonists, activating the cyclic GMP-AMP synthase (cGAS)-STING pathway in cancer cells and tumour-associated macrophages (TAMs). Loading engineered OMVs with photoimmunotherapy photosensitisers further enhanced tumour inhibition and STING activation in TAMs. Combining nanobody-engineered OMV-mediated photoimmunotherapy with CD47 blockade effectively suppressed primary and metastatic tumours, establishing sustained antitumour immune memory. This study demonstrates the potential of nanobody-engineered OMVs as STING agonists and provides insights into novel OMV-based immunotherapeutic strategies harnessing the innate immune system against cancer. Our findings open new avenues for OMV applications in tumour immunotherapy, offering a promising approach to overcome current limitations in cancer treatment.
Keywords: CDH17; STING pathway; bacterial outer membrane vesicle; nanobody; photoimmunotherapy; tumour‐associated macrophages.
© 2025 The Author(s). Journal of Extracellular Vesicles published by Wiley Periodicals, LLC on behalf of the International Society for Extracellular Vesicles.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures


Similar articles
-
Biomineralized Engineered Bacterial Outer Membrane Vesicles as cGAS-STING Nanoagonists Synergize with Lactate Metabolism Modulation to Potentiate Immunotherapy.J Am Chem Soc. 2025 Jul 16;147(28):24555-24572. doi: 10.1021/jacs.5c05148. Epub 2025 Jul 2. J Am Chem Soc. 2025. PMID: 40601938
-
Engineered bacterial membrane vesicle as safe and efficient nano-heaters to reprogram tumor microenvironment for enhanced immunotherapy.J Control Release. 2024 Oct;374:127-139. doi: 10.1016/j.jconrel.2024.08.008. Epub 2024 Aug 13. J Control Release. 2024. PMID: 39122216
-
Engineered Outer Membrane Vesicles as Nanosized Immune Cell Engagers for Enhanced Solid Tumor Immunotherapy.ACS Nano. 2024 Nov 5;18(44):30332-30344. doi: 10.1021/acsnano.4c07364. Epub 2024 Oct 25. ACS Nano. 2024. PMID: 39454084
-
Engineering Versatile Bacteria-Derived Outer Membrane Vesicles: An Adaptable Platform for Advancing Cancer Immunotherapy.Adv Sci (Weinh). 2024 Sep;11(33):e2400049. doi: 10.1002/advs.202400049. Epub 2024 Jul 1. Adv Sci (Weinh). 2024. PMID: 38952055 Free PMC article. Review.
-
Clinical applications of STING agonists in cancer immunotherapy: current progress and future prospects.Front Immunol. 2024 Oct 2;15:1485546. doi: 10.3389/fimmu.2024.1485546. eCollection 2024. Front Immunol. 2024. PMID: 39421752 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
- 82373775/National Natural Science Foundation of China
- 82403904/National Natural Science Foundation of China
- 82304518/National Natural Science Foundation of China
- 82400727/National Natural Science Foundation of China
- 2022YFC2303600/National Key Research and Development Program of China
- 2023YFE0204500/The Key Special Project of Strategic Science and Technology Innovation Cooperation from National Key R&D Program (2023YFE0204500)
- BX20240271/The China National Postdoctoral Program for Innovative Talents
- 2023M742698/China Postdoctoral Science Foundation
- GZC20241274/China Postdoctoral Science Foundation
- 413000553/The Fundamental Research Funds for the Central Universities
- 2024AFB163/The Hubei Natural Science Foundation
- 2004HBBHJD082/The Hubei Postdoctoral Science Foundation
- 2004HBBHJD058/The Hubei Postdoctoral Science Foundation
- D2403013/The Shenzhen Medical Research Fund
- B2302051/The Shenzhen Medical Research Fund
- CI2023D003/The Scientific and Technological Innovation Project of China Academy of Chinese Medical Sciences
- CI2021B014/The Scientific and Technological Innovation Project of China Academy of Chinese Medical Sciences
- CI2023D008/The Scientific and Technological Innovation Project of China Academy of Chinese Medical Sciences
- CI2023E002/The CACMS Innovation Fund
- CI2021A05101/The CACMS Innovation Fund
- CI2021A05104/The CACMS Innovation Fund
- RCBS20210706092213007/The Shenzhen Science and Technology Innovation Committee
- RCYX20221008092950121/The Shenzhen Science and Technology Innovation Committee
- JCYJ20200109120205924/The Shenzhen Science and Technology Innovation Committee
- GDRC20212/The Natural Science Foundation of Top Talent of SZTU
- SZXK046/Shenzhen Key Medical Discipline Construction Fund
- GJHZ20240218114508015/International Science and Technology Cooperation for Shenzhen Technology Innovation Plan
- KCXFZ20201221173612034/Shenzhen Governmental Sustainable Development Fund
- ZDSYS201504301616234/Shenzhen key Laboratory of Kidney Diseases
- SZGSP001/Shenzhen Fund for Guangdong Provincial High-level Clinical Key Specialties
- SYWGSCGZH202405/Shenzhen People's Hospital Fund
- ZLYNXM202004/The Cancer Research and Translational Platform Project of Zhongnan Hospital of Wuhan University
- PTXM2023008/The medical Sci-Tech innovation platform of Zhongnan Hospital of Wuhan University
- PTXM2024029/The medical Sci-Tech innovation platform of Zhongnan Hospital of Wuhan University
- ZNYB2023002/The Program of Excellent Doctor of Zhongnan Hospital of Wuhan University
- ZNYB20240015/The Program of Excellent Doctor of Zhongnan Hospital of Wuhan University
LinkOut - more resources
Full Text Sources
Medical
Research Materials
