

The Pathologic Actions of Phosphate in CKD
- PMID: 40241437
- PMCID: PMC12233857
- DOI: 10.34067/KID.0000000820
The Pathologic Actions of Phosphate in CKD
Abstract
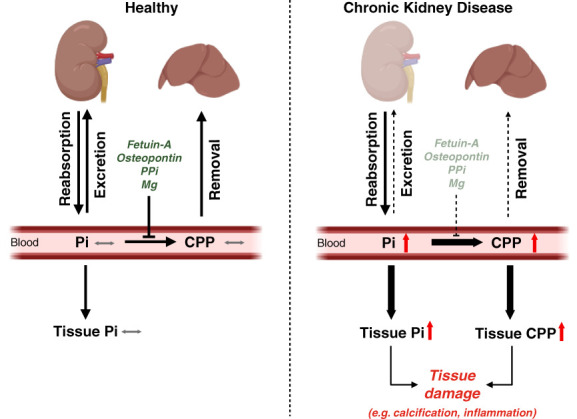
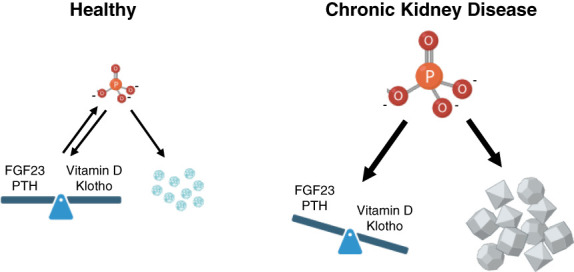
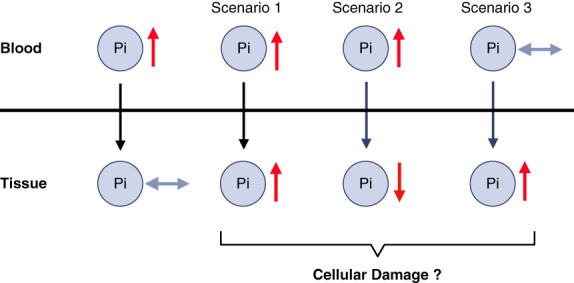
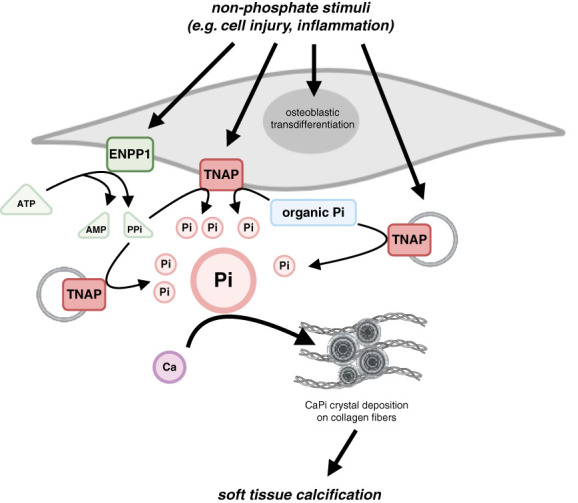
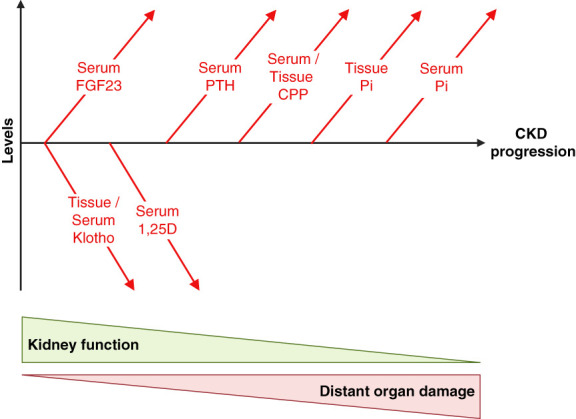
CKD is associated with high serum levels of phosphate (also called hyperphosphatemia), which is a main driver of soft tissue calcifications and potentially other pathologic changes that are associated with CKD. However, it remains unclear in what form and through which mechanisms and targets elevated phosphate can damage cells and tissues. Rises in serum phosphate levels are accompanied by changes in the endocrine regulators of phosphate metabolism and result in the formation of calcium-phosphate crystals, and all three events can have pathologic actions on various tissues. Furthermore, tissues can accumulate phosphate from the circulation, and cells can generate free phosphate in their environment independently from circulating phosphate, which both result in local elevations of phosphate that could also contribute to tissue damage. It is important to better understand the various scenarios underlying the pathologic actions of hyperphosphatemia, as some of them suggest that measuring extracellular serum phosphate, which is the gold standard to estimate overall phosphate status of the body, is not sufficient to do so. Understanding the pathologic actions of phosphate on a conceptual level should not only help to design more efficient detection tools for phosphate but also to identify phosphate-induced pathomechanisms which could provide us with novel drug targets to tackle phosphate-driven pathologies in CKD. Here, we discuss the different concepts and scenarios that could underlie the widespread pathologic actions of hyperphosphatemia in CKD.
Trial registration: ClinicalTrials.gov NCT04095039 NCT03573089.
Keywords: CKD; hyperphosphatemia; vascular calcification.
Copyright © 2025 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Society of Nephrology.
Conflict of interest statement
Disclosure forms, as provided by each author, are available with the online version of the article at
Figures
Similar articles
-
Soft tissue calcifications in chronic kidney disease-beyond the vasculature.Pflugers Arch. 2025 Aug;477(8):1037-1059. doi: 10.1007/s00424-025-03098-0. Epub 2025 Jun 5. Pflugers Arch. 2025. PMID: 40471241 Free PMC article. Review.
-
Elevated phosphate levels in CKD - a direct threat for the heart.Nephrol Dial Transplant. 2025 Jun 30;40(7):1294-1309. doi: 10.1093/ndt/gfaf001. Nephrol Dial Transplant. 2025. PMID: 39809572 Review.
-
Chronic Kidney Disease-associated Lung Injury Is Mediated by Phosphate-induced MAPK/AKT Signaling.Am J Respir Cell Mol Biol. 2024 Dec;71(6):659-676. doi: 10.1165/rcmb.2024-0008OC. Am J Respir Cell Mol Biol. 2024. PMID: 39088759 Free PMC article.
-
Comparison of Two Modern Survival Prediction Tools, SORG-MLA and METSSS, in Patients With Symptomatic Long-bone Metastases Who Underwent Local Treatment With Surgery Followed by Radiotherapy and With Radiotherapy Alone.Clin Orthop Relat Res. 2024 Dec 1;482(12):2193-2208. doi: 10.1097/CORR.0000000000003185. Epub 2024 Jul 23. Clin Orthop Relat Res. 2024. PMID: 39051924
-
The Black Book of Psychotropic Dosing and Monitoring.Psychopharmacol Bull. 2024 Jul 8;54(3):8-59. Psychopharmacol Bull. 2024. PMID: 38993656 Free PMC article. Review.
Cited by
-
Soft tissue calcifications in chronic kidney disease-beyond the vasculature.Pflugers Arch. 2025 Aug;477(8):1037-1059. doi: 10.1007/s00424-025-03098-0. Epub 2025 Jun 5. Pflugers Arch. 2025. PMID: 40471241 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
