Rapid discovery of functional RNA domains
- PMID: 40243058
- PMCID: PMC12004112
- DOI: 10.1093/nar/gkaf307
Rapid discovery of functional RNA domains
Abstract
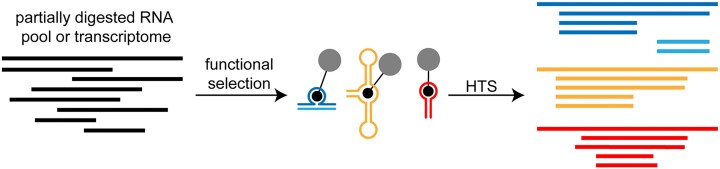
Many strategies have been implemented to enrich an RNA population for a selectable function, but demarcation of the optimal functional motifs or minimal structures within longer libraries remains a lengthy and tedious process. To overcome this problem, we have developed a technique that isolates minimal active segments from complex heterogeneous pools of RNAs. This method allows for truncations to occur at both 5' and 3' ends of functional domains and introduces independent primer-binding sequences, thereby removing sequence and structure bias introduced by constant-sequence regions. We show examples of minimization for genomic and synthetic aptamers and demonstrate that the method can directly reveal an active RNA assembled from multiple strands, facilitating the development of heterodimeric structures used in cellular sensors. This approach provides a pipeline to experimentally define the boundaries of active domains and accelerate the discovery of functional RNAs.
© The Author(s) 2025. Published by Oxford University Press on behalf of Nucleic Acids Research.
Conflict of interest statement
None declared.
Figures