

Application of Density Functional Theory to Molecular Engineering of Pharmaceutical Formulations
- PMID: 40244098
- PMCID: PMC11989887
- DOI: 10.3390/ijms26073262
Application of Density Functional Theory to Molecular Engineering of Pharmaceutical Formulations
Abstract
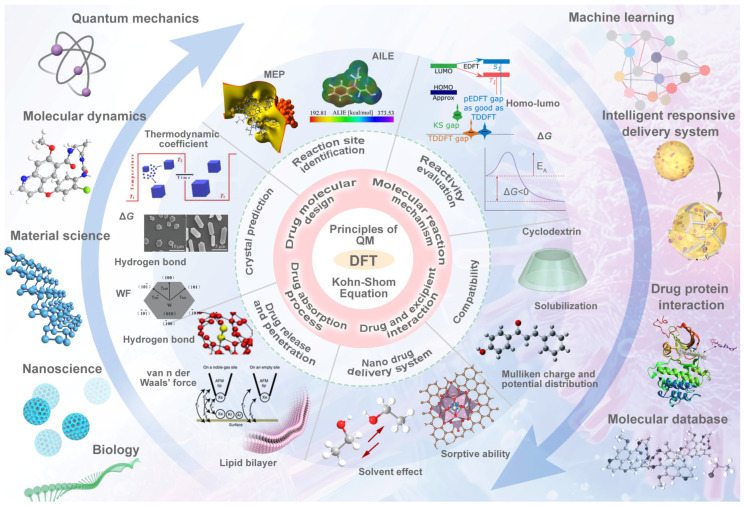
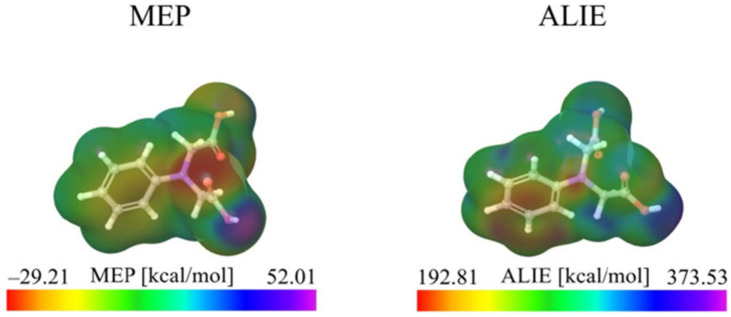
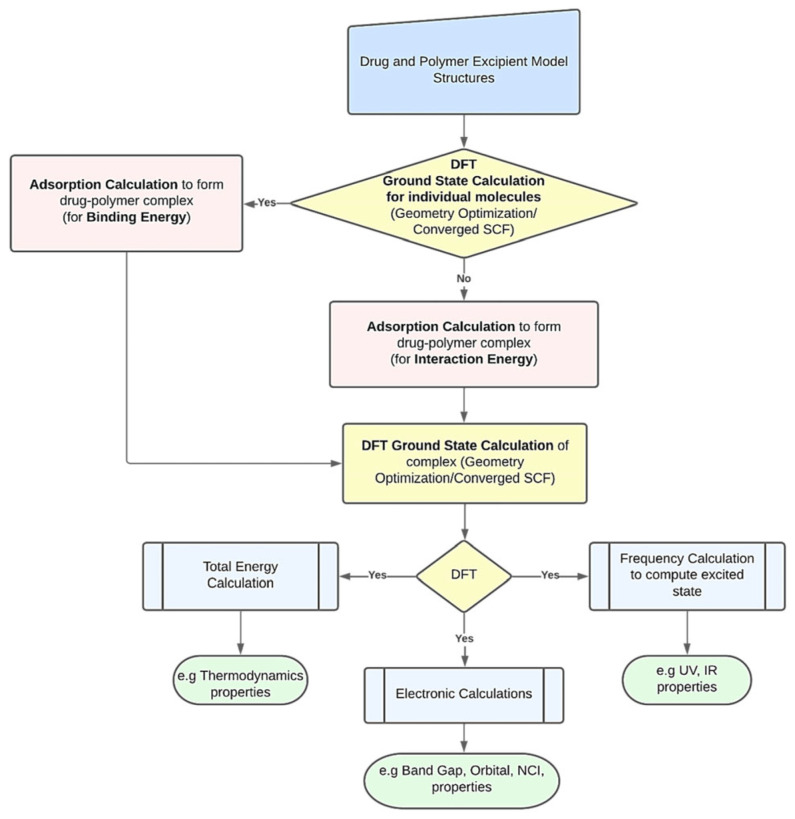
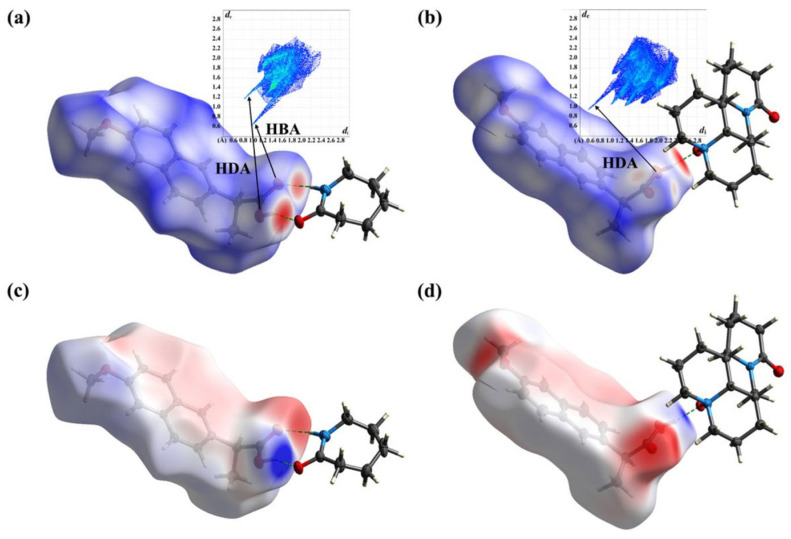
This review systematically examines the pivotal applications of the Density Functional Theory (DFT) in drug formulation design, emphasizing its capability to elucidate molecular interaction mechanisms through quantum mechanical calculations. By solving the Kohn-Sham equations with precision up to 0.1 kcal/mol, DFT enables accurate electronic structure reconstruction, providing theoretical guidance for optimizing drug-excipient composite systems. In solid dosage forms, DFT clarifies the electronic driving forces governing active pharmaceutical ingredient (API)-excipient co-crystallization, predicting reactive sites and guiding stability-oriented co-crystal design. For nanodelivery systems, DFT optimizes carrier surface charge distribution through van der Waals interactions and π-π stacking energy calculations, thereby enhancing targeting efficiency. Furthermore, DFT combined with solvation models (e.g., COSMO) quantitatively evaluates polar environmental effects on drug release kinetics, delivering critical thermodynamic parameters (e.g., ΔG) for controlled-release formulation development. Notably, DFT-driven co-crystal thermodynamic analysis and pH-responsive release mechanism modeling substantially reduce experimental validation cycles. While DFT faces challenges in dynamic simulations of complex solvent environments, its integration with molecular mechanics and multiscale frameworks has achieved computational breakthroughs. This work offers interdisciplinary methodology support for accelerating data-driven formulation design.
Keywords: density functional theory; drug release; molecular interactions; pharmaceutical formulations.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Combining crystalline and polymeric excipients in API solid dispersions - Opportunity or risk?Eur J Pharm Biopharm. 2021 Jan;158:323-335. doi: 10.1016/j.ejpb.2020.11.025. Epub 2020 Dec 6. Eur J Pharm Biopharm. 2021. PMID: 33296719
-
Pharmaceutical excipients in pediatric and geriatric drug formulations: safety, efficacy, and regulatory perspectives.Pharm Dev Technol. 2025 Jan;30(1):1-9. doi: 10.1080/10837450.2024.2441181. Epub 2024 Dec 14. Pharm Dev Technol. 2025. PMID: 39660788 Review.
-
Solubility Enhancement of Active Pharmaceutical Ingredients through Liquid Hydrotrope Addition: A Thermodynamic Analysis.Mol Pharm. 2025 Mar 3;22(3):1408-1418. doi: 10.1021/acs.molpharmaceut.4c01117. Epub 2025 Feb 13. Mol Pharm. 2025. PMID: 39945746 Free PMC article.
-
Navigating the Solution to Drug Formulation Problems at Research and Development Stages by Amorphous Solid Dispersion Technology.Recent Adv Drug Deliv Formul. 2024;18(2):79-99. doi: 10.2174/0126673878271641231201065151. Recent Adv Drug Deliv Formul. 2024. PMID: 38062659 Review.
-
Investigation of Drug-Excipient Interactions in Biclotymol Amorphous Solid Dispersions.Mol Pharm. 2018 Mar 5;15(3):1112-1125. doi: 10.1021/acs.molpharmaceut.7b00993. Epub 2018 Jan 29. Mol Pharm. 2018. PMID: 29328661
Cited by
-
Crystallographic and DFT study of novel dimethoxybenzene derivatives.BMC Chem. 2025 May 14;19(1):127. doi: 10.1186/s13065-025-01496-0. BMC Chem. 2025. PMID: 40369581 Free PMC article.
-
A review of herbal therapeutics for the prevention and management of poxvirus infections.Arch Microbiol. 2025 Jun 25;207(8):186. doi: 10.1007/s00203-025-04367-3. Arch Microbiol. 2025. PMID: 40560228 Review.
-
Identification of Hydroxyl and Polysiloxane Compounds via Infrared Absorption Spectroscopy with Targeted Noise Analysis.Polymers (Basel). 2025 May 30;17(11):1533. doi: 10.3390/polym17111533. Polymers (Basel). 2025. PMID: 40508775 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
