

Quantitative Changes in the Proteome of Chronically Inflamed Lacrimal Glands From a Sjögren's Disease Animal Model
- PMID: 40244610
- PMCID: PMC12013672
- DOI: 10.1167/iovs.66.4.44
Quantitative Changes in the Proteome of Chronically Inflamed Lacrimal Glands From a Sjögren's Disease Animal Model
Abstract
Purpose: The lacrimal gland (LG) is the major source of aqueous tears, and insufficient LG secretion leads to aqueous-deficient dry eye (ADDE) disease. To provide a foundational description of LG's protein expression patterns, we prepared protein extracts of LGs from a wild-type and an ADDE mouse model and analyzed the proteome by quantitative mass spectrometry.
Methods: LGs were isolated from an ADDE mouse model, male non-obese diabetic (NOD) mice and control wild-type BALB/c mice (n = 6 each). Protein samples were prepared in urea-based lysis buffer and protein concentrations determined by the BCA method. The equivalent of 200 µg protein were tryptically digested and analyzed by nanoflow liquid chromatography tandem mass spectrometry (LC-MS/MS). Proteins were identified and quantified using the PEAKS X bioinformatics suite. Downstream differential protein expression analysis was performed using the MS-DAP R package. Selected significantly differentially expressed and detected proteins were subjected to spatial expression analysis using immunohistochemistry.
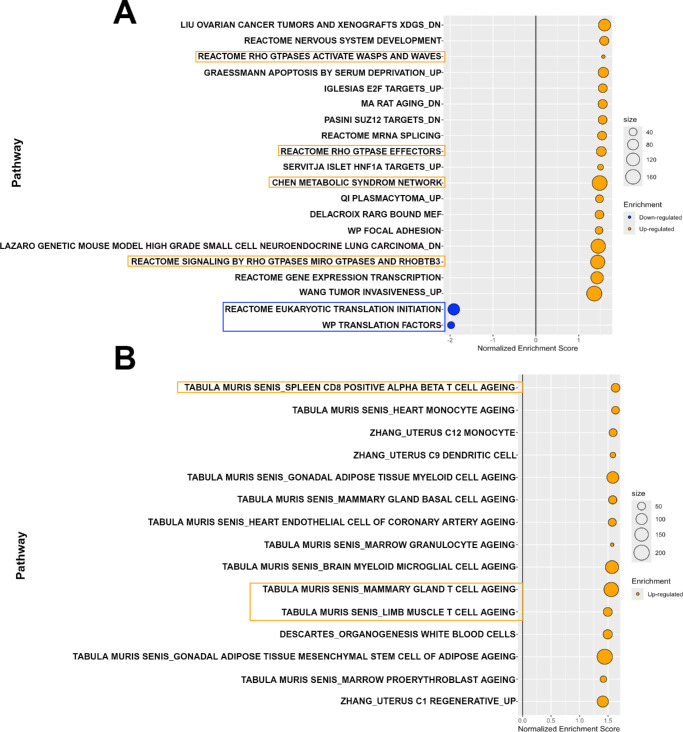
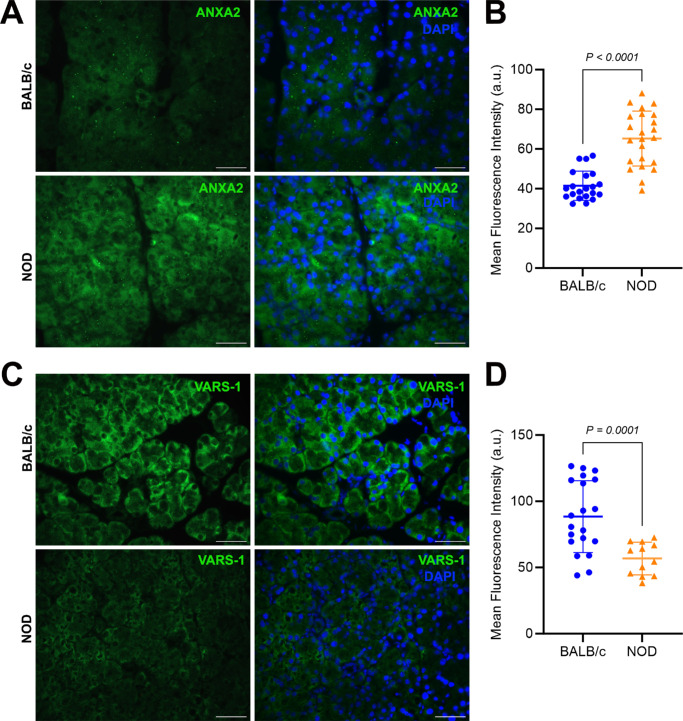
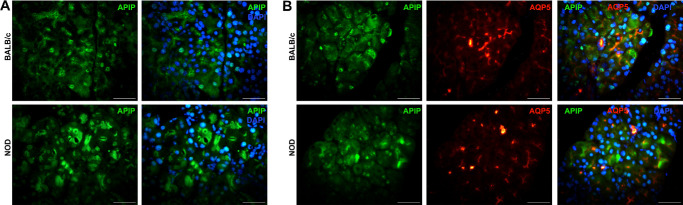
Results: Cumulatively, the LC-MS/MS-based proteomics analyses of the murine LG samples identified a total of 31,932 peptide sequences resulting in 2617 protein identifications at a 1% false discovery rate at the peptide and protein level. Principal component analysis (PCA) and hierarchical cluster analysis revealed a separation of NOD and BALB/c samples. Overall, protein diversity was consistently higher in NOD samples. After applying global peptide filter criteria and peptide-to-protein rollup, 1750 remaining proteins were subjected to differential expression analysis using the MSqRob algorithm, which identified 580 proteins with statistically significant expression differences. Data are available via ProteomeXchange with identifier PXD060937. At the cellular level, the up- and downregulation of select proteins were confirmed by immunohistochemistry.
Conclusions: Our data suggest that chronic inflammation leads to significant alterations in the LG proteome. Ongoing studies aim to identify potentially unique, inflammation-induced proteins that could be amenable to pharmacological modulation.
Conflict of interest statement
Disclosure:
Figures





Similar articles
-
Proteomic analysis revealed the altered tear protein profile in a rabbit model of Sjögren's syndrome-associated dry eye.Proteomics. 2013 Aug;13(16):2469-81. doi: 10.1002/pmic.201200230. Proteomics. 2013. PMID: 23733261 Free PMC article.
-
Altered distribution of aquaporin 5 and its C-terminal binding protein in the lacrimal glands of a mouse model for Sjögren's syndrome.Curr Eye Res. 2008 Aug;33(8):621-9. doi: 10.1080/02713680802262819. Curr Eye Res. 2008. PMID: 18696337
-
Inhibition of Cathepsin S Reduces Lacrimal Gland Inflammation and Increases Tear Flow in a Mouse Model of Sjögren's Syndrome.Sci Rep. 2019 Jul 2;9(1):9559. doi: 10.1038/s41598-019-45966-7. Sci Rep. 2019. PMID: 31267034 Free PMC article.
-
Review of genes potentially related to hyposecretion in male non-obese diabetic (NOD) mice, a Sjögren's syndrome model.J Oral Biosci. 2023 Sep;65(3):211-217. doi: 10.1016/j.job.2023.05.001. Epub 2023 May 18. J Oral Biosci. 2023. PMID: 37209839 Review.
-
Fas (APO-1/CD95)-assisted suicide in NOD exocrine glands.Clin Exp Rheumatol. 1998 Nov-Dec;16(6):659-61. Clin Exp Rheumatol. 1998. PMID: 9844755 Review. No abstract available.
References
-
- Hodges RR, Dartt DA.. Regulatory pathways in lacrimal gland epithelium. Int Rev Cytol. 2003; 231: 129–196. - PubMed
-
- Barabino S, Aragona P, di Zazzo A, Rolando M. Updated definition and classification of dry eye disease: renewed proposals using the nominal group and Delphi techniques. Eur J Ophthalmol. 2021; 31: 42–48. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
