

Bi-allelic pathogenic variants in TRMT1 disrupt tRNA modification and induce a neurodevelopmental disorder
- PMID: 40245862
- PMCID: PMC12120178
- DOI: 10.1016/j.ajhg.2025.03.015
Bi-allelic pathogenic variants in TRMT1 disrupt tRNA modification and induce a neurodevelopmental disorder
Abstract
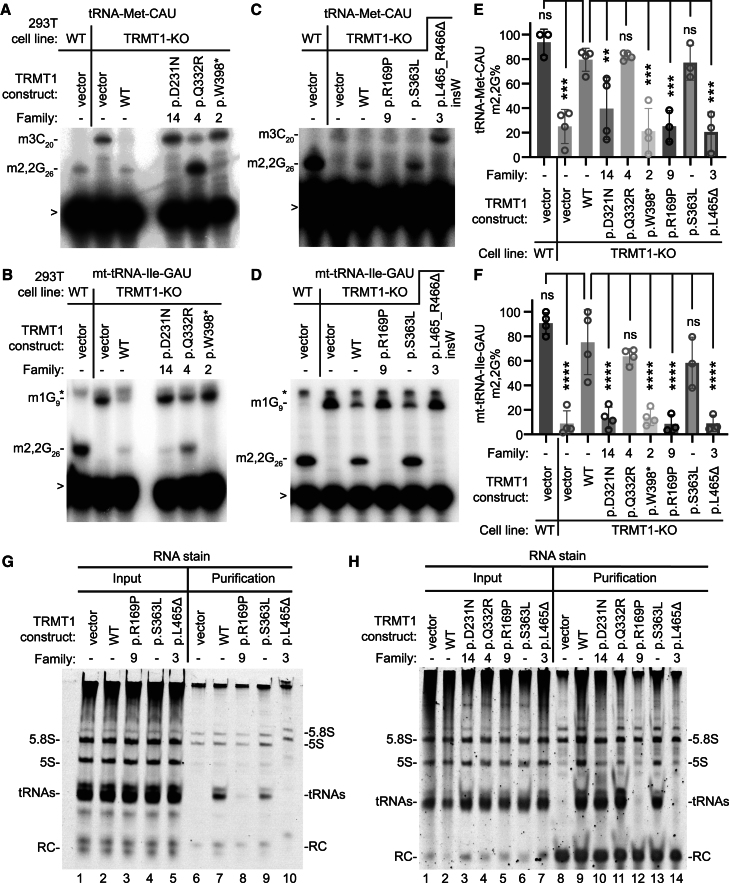
The post-transcriptional modification of tRNAs plays a crucial role in tRNA structure and function. Pathogenic variants in tRNA-modification enzymes have been implicated in a wide range of human neurodevelopmental and neurological disorders. However, the molecular basis for many of these disorders remains unknown. Here, we describe a comprehensive cohort of 43 individuals from 31 unrelated families with bi-allelic variants in tRNA methyltransferase 1 (TRMT1). These individuals present with a neurodevelopmental disorder universally characterized by developmental delay and intellectual disability, accompanied by variable behavioral abnormalities, epilepsy, and facial dysmorphism. The identified variants include ultra-rare TRMT1 variants, comprising missense and predicted loss-of-function variants, which segregate with the observed clinical pathology. Our findings reveal that several variants lead to mis-splicing and a consequent loss of TRMT1 protein accumulation. Moreover, cells derived from individuals harboring TRMT1 variants exhibit a deficiency in tRNA modifications catalyzed by TRMT1. Molecular analysis reveals distinct regions of TRMT1 required for tRNA-modification activity and binding. Notably, depletion of Trmt1 protein in zebrafish is sufficient to induce developmental and behavioral phenotypes along with gene-expression changes associated with disrupted cell cycle, immune response, and neurodegenerative disorders. Altogether, these findings demonstrate that loss of TRMT1-catalyzed tRNA modifications leads to intellectual disability and provides insight into the molecular underpinnings of tRNA-modification deficiency caused by pathogenic TRMT1 variants.
Keywords: TRMT1; disease model; intellectual disability; neurodevelopmental disorder; tRNA modification; zebrafish.
Copyright © 2025 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests M.M.M. and D.A.C. are employees of and may own stock in GeneDx, LLC. R.S. is on the advisory board of Guide Genetics and Egetis Pharmaceuticals.
Figures






References
-
- Daily D.K., Ardinger H.H., Holmes G.E. Identification and evaluation of mental retardation. Am. Fam. Physician. 2000;61:1059–1070. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
