

Investigation of an oncolytic herpes simplex virus as a potential therapeutic agent for gastroenteropancreatic neuroendocrine neoplasms
- PMID: 40247049
- PMCID: PMC12006505
- DOI: 10.1038/s41598-025-98588-7
Investigation of an oncolytic herpes simplex virus as a potential therapeutic agent for gastroenteropancreatic neuroendocrine neoplasms
Abstract
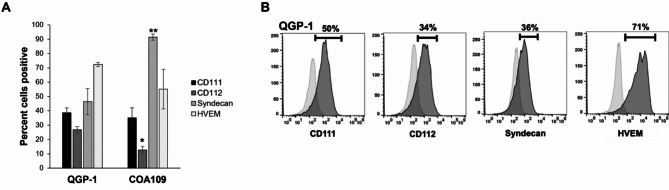
Gastroenteropancreatic neuroendocrine tumors (GEP-NETs) present unique challenges due to their heterogeneity and limited treatment options. Oncolytic virotherapy has emerged as a promising therapeutic for other NETs and thus, we sought to investigate the potential of an engineered oncolytic herpes simplex virus (oHSV), M002, for GEP-NETS. We employed an established long-term passage GEP-NET cell line and a unique, human pediatric patient-derived xenograft GEP-NET line. We found the virus to effectively infect, replicate within, and kill both cell lines in vitro. Similar effects were noted in vivo, with M002 decreasing tumor growth and improving overall survival in mice bearing tumors from both the established cell line and human GEP-NET PDX. Overall, these studies provide an evaluation of an oncolytic HSV in GEP-NETs, highlighting its therapeutic potential and considerations for clinical translation.
Keywords: Neuroendocrine tumors; Oncolytic virus; Patient derived xenografts; Translational research.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: Dr James M. Markert holds equity (< 8%) in Aettis, Inc. (a company that holds stocks of oncolytic virus); Treovir, Inc (25%), a company holding intellectual property and funding clinical trials of oncolytic virus for pediatric brain tumors. A company that Dr J.M. Markert formerly held equity in (< 8%) Catherex, Inc., was purchased in a structured buyout. Dr J.M. Markert has served as a consultant for Imugene. He also holds a fraction of the IP associated with oncolytic virus C134, which is licensed by Mustang Biotech. The following authors have no competing interest: Colin H. Quinn, Janet R. Julson, Michael H. Erwin, Hooper R. Markert, Laura V. Bownes, Jerry E. Stewart, Sorina Shirley, Karina J. Yoon, Jamie M. Aye, and Elizabeth A. Beierle.
Figures




References
-
- Sarvida, M. E. & O’Dorisio, M. S. Neuroendocrine tumors in children and young adults: rare or not so rare. Endocrinol. Metab. Clin. North Am.40(1), 65–80 (2011). - PubMed
-
- Zhuge, Y. et al. Pediatric intestinal foregut and small bowel solid tumors: A review of 105 cases. J. Surg. Res.156(1), 95–102 (2009). - PubMed
-
- Haddad, T., Fard-Esfahani, A. & Vali, R. A review of pediatric neuroendocrine tumors, their detection, and treatment by radioisotopes. Nucl. Med. Commun.42(1), 21–31 (2021). - PubMed
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
