

Interplay of genetic and clinical factors in cancer-associated thrombosis: Deciphering the prothrombotic landscape of colorectal cancer
- PMID: 40248375
- PMCID: PMC12001197
- DOI: 10.3748/wjg.v31.i14.103901
Interplay of genetic and clinical factors in cancer-associated thrombosis: Deciphering the prothrombotic landscape of colorectal cancer
Abstract
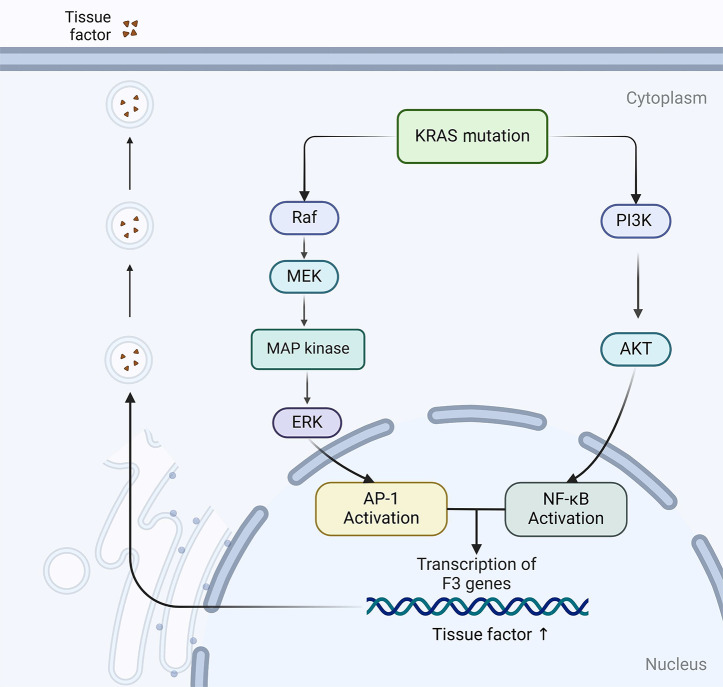
Colorectal cancer (CRC), the third most prevalent cancer globally, exhibits a notable association with venous thromboembolism (VTE), significantly impacting patient morbidity and mortality. We delve into the complex pathogenesis of cancer-associated thrombosis (CAT) in CRC, highlighting the interplay of clinical risk factors and tumor-specific mechanisms. Our comprehensive review synthesizes the current understanding of CRC's pro-thrombotic tendencies, examining both general clinical factors (e.g., age, gender, obesity, prior VTE history) and tumor-specific aspects (e.g., tumor location, stage, targeted therapies). Key findings illustrate how CRC cells themselves actively contribute to coagulation cascade activation through various procoagulant elements such as tissue factor, cancer procoagulant, and extracellular vesicles. We also explore how CRC influences host cells to adopt a procoagulant phenotype, thereby exacerbating thrombotic risks. This review underscores the role of genetic mutations in CRC (e.g., KRAS, p53) in modulating coagulation-related protein expression and thrombosis risks. An in-depth understanding of the genetic landscape specific to CRC subtypes is essential for developing targeted anticoagulation strategies and could significantly advance thrombosis prevention while improving the overall management of patients with CRC. This highlights the urgent need for precision in addressing CAT within clinical settings.
Keywords: Cancer-associated thrombosis; Colorectal cancer cell; Platelet; Tissue factor; Venous thromboembolism.
©The Author(s) 2025. Published by Baishideng Publishing Group Inc. All rights reserved.
Conflict of interest statement
Conflict-of-interest statement: All the authors report no relevant conflicts of interest for this article.
Figures



References
-
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71:209–249. - PubMed
-
- Falanga A, Russo L. Epidemiology, risk and outcomes of venous thromboembolism in cancer. Hamostaseologie. 2012;32:115–125. - PubMed
-
- Heit JA, O'Fallon WM, Petterson TM, Lohse CM, Silverstein MD, Mohr DN, Melton LJ 3rd. Relative impact of risk factors for deep vein thrombosis and pulmonary embolism: a population-based study. Arch Intern Med. 2002;162:1245–1248. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
