

SOI: robust identification of orthologous synteny with the Orthology Index and broad applications in evolutionary genomics
- PMID: 40248914
- PMCID: PMC12006799
- DOI: 10.1093/nar/gkaf320
SOI: robust identification of orthologous synteny with the Orthology Index and broad applications in evolutionary genomics
Abstract
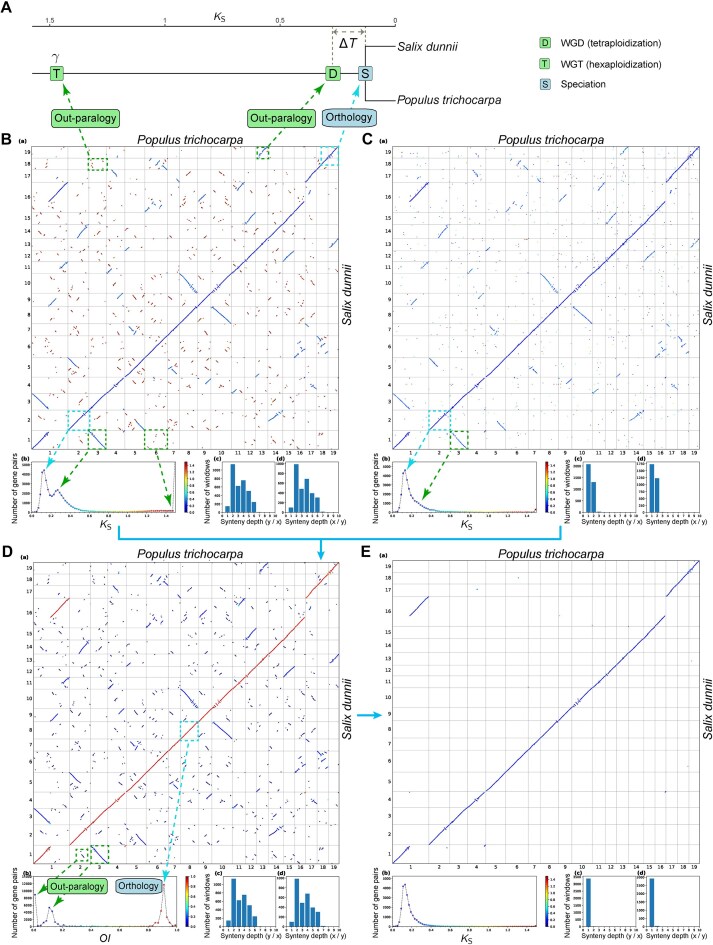
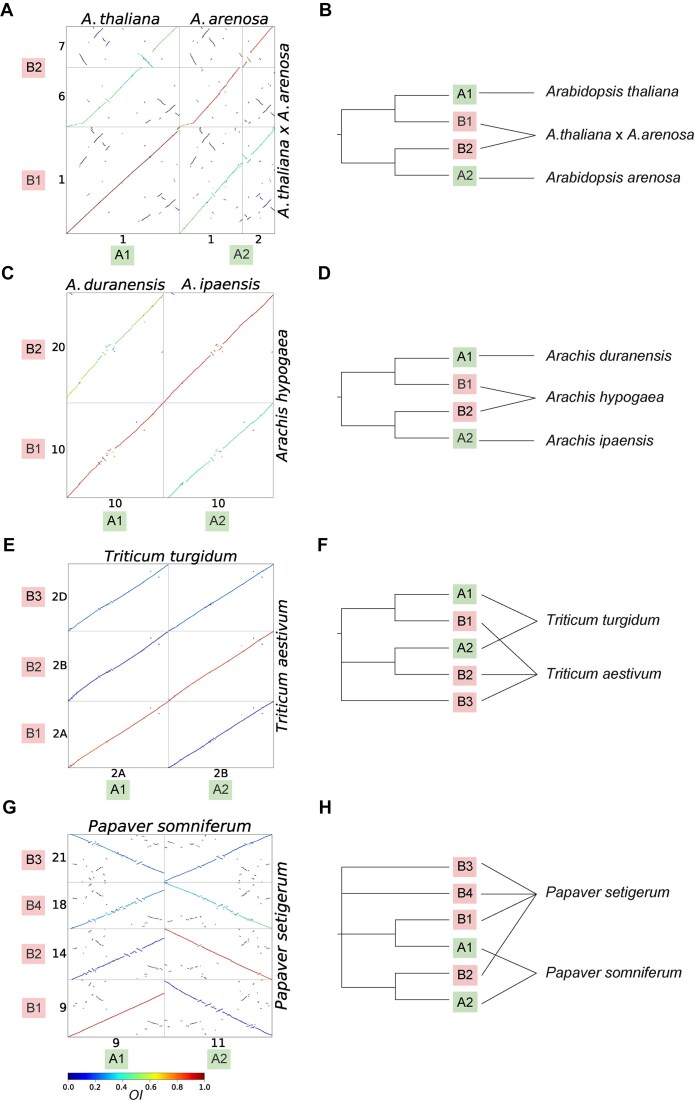
With the explosive growth of whole-genome datasets, accurate detection of orthologous synteny has become crucial for reconstructing evolutionary history. However, current methods for identifying orthologous synteny face great limitations, particularly in scaling with varied polyploidy histories and accurately removing out-paralogous synteny. In this study, we developed a scalable and robust approach, based on the Orthology Index (OI), to effectively identify orthologous synteny. Our evaluation across a large-scale empirical dataset with diverse polyploidization events demonstrated the high reliability and robustness of the OI method. Simulation-based benchmarks further validated the accuracy of our method, showing its superior performance against existing methods across a wide range of scenarios. Additionally, we explored its broad applications in reconstructing the evolutionary histories of plant genomes, including the inference of polyploidy, identification of reticulation, and phylogenomics. In conclusion, OI offers a robust, interpretable, and scalable approach for identifying orthologous synteny, facilitating more accurate and efficient analyses in plant evolutionary genomics.
© The Author(s) 2025. Published by Oxford University Press on behalf of Nucleic Acids Research.
Conflict of interest statement
None declared.
Figures




References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
