

Transcriptomic profiling of severe and critical COVID-19 patients reveals alterations in expression, splicing and polyadenylation
- PMID: 40251257
- PMCID: PMC12008264
- DOI: 10.1038/s41598-025-95905-y
Transcriptomic profiling of severe and critical COVID-19 patients reveals alterations in expression, splicing and polyadenylation
Abstract
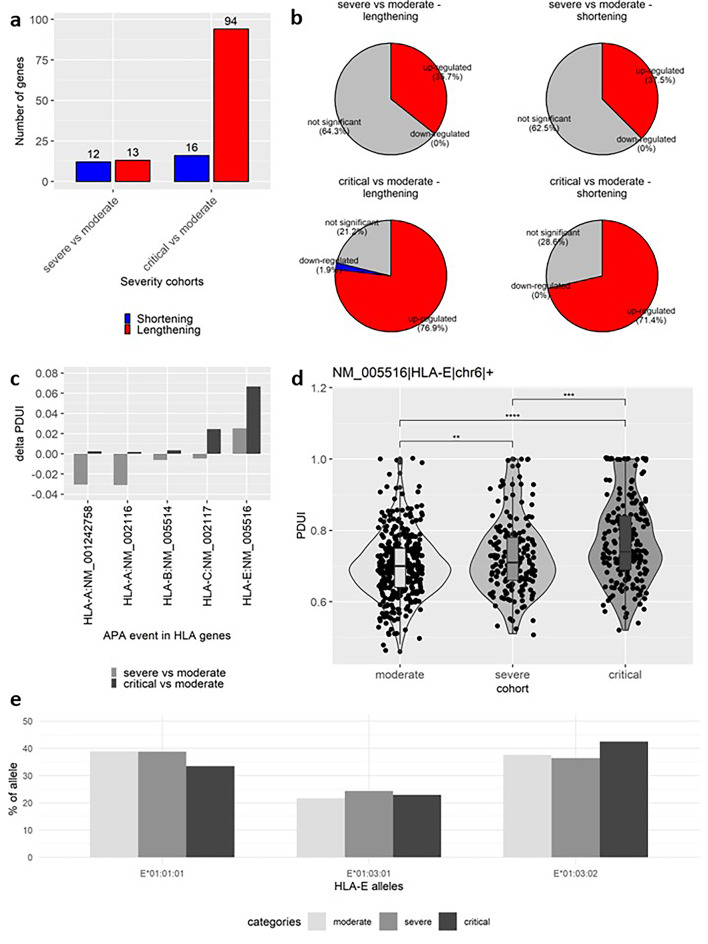
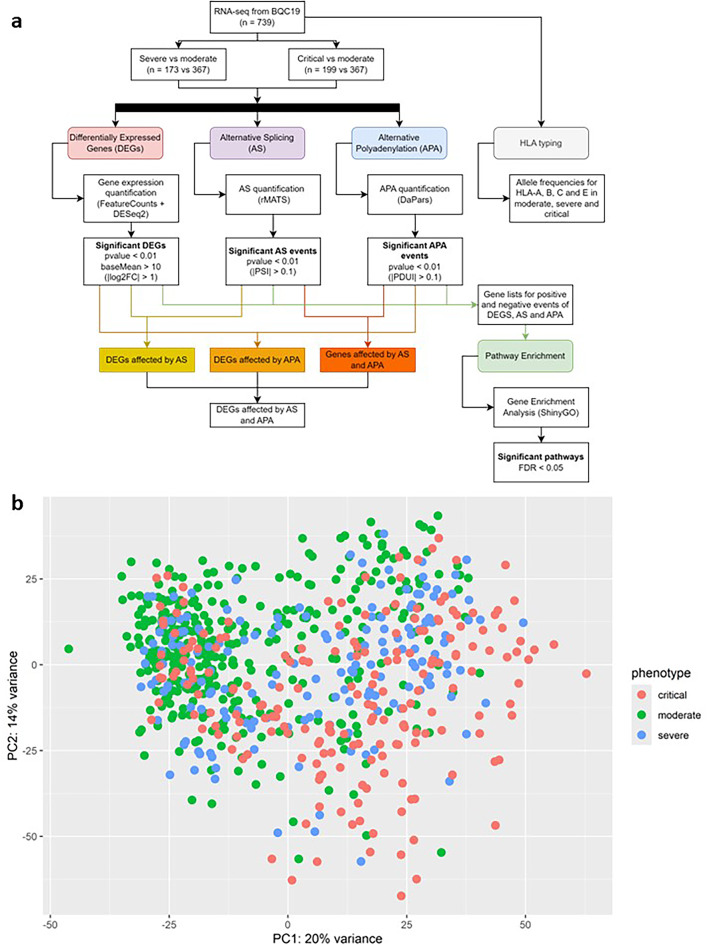
Coronavirus disease 2019 (COVID-19) is a multi-systemic illness that became a pandemic in March 2020. Although environmental factors and comorbidities can influence disease progression, there is a lack of prognostic markers to predict the severity of COVID-19 illness. Identifying these markers is crucial for improving patient outcomes and appropriately allocating scarce resources. Here, an RNA-sequencing study was conducted on blood samples from unvaccinated, hospitalized patients divided by disease severity; 367 moderate, 173 severe, and 199 critical. Using a bioinformatics approach, we identified differentially expressed genes (DEGs), alternative splicing (AS) and alternative polyadenylation (APA) events that were severity-dependent. In the severe group, we observed a higher expression of kappa immunoglobulins compared to the moderate group. In the critical cohort, a majority of AS events were mutually exclusive exons and APA genes mostly had longer 3'UTRs. Interestingly, multiple genes associated with cytoskeleton, TUBA4A, NRGN, BSG, and CD300A, were differentially expressed, alternatively spliced and polyadenylated in the critical group. Furthermore, several inflammation-related pathways were observed predominantly in critical vs. moderate. We demonstrate that integrating multiple downstream analyses of transcriptomics, from moderate, severe, and critical patients confers a significant advantage in identifying relevant dysregulated genes and pathways.
Keywords: Alternative polyadenylation; Alternative splicing; COVID-19; Differentially expressed genes; Pathway enrichment; Transcriptomics.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests.
Figures



Similar articles
-
Genome-Wide Profiling Reveals Alternative Polyadenylation of Innate Immune-Related mRNA in Patients With COVID-19.Front Immunol. 2021 Oct 27;12:756288. doi: 10.3389/fimmu.2021.756288. eCollection 2021. Front Immunol. 2021. PMID: 34777369 Free PMC article.
-
Comparative Transcriptomic Analyses of Peripheral Blood Mononuclear Cells of COVID-19 Patients without Pneumonia and with Severe Pneumonia in the First Year of Follow-Up.Viruses. 2024 Jul 28;16(8):1211. doi: 10.3390/v16081211. Viruses. 2024. PMID: 39205185 Free PMC article.
-
MixOmics Integration of Biological Datasets Identifies Highly Correlated Variables of COVID-19 Severity.Int J Mol Sci. 2025 May 15;26(10):4743. doi: 10.3390/ijms26104743. Int J Mol Sci. 2025. PMID: 40429886 Free PMC article.
-
3'UTR Diversity: Expanding Repertoire of RNA Alterations in Human mRNAs.Mol Cells. 2023 Jan 31;46(1):48-56. doi: 10.14348/molcells.2023.0003. Epub 2023 Jan 20. Mol Cells. 2023. PMID: 36697237 Free PMC article. Review.
-
Alternative polyadenylation analysis in animals and plants: newly developed strategies for profiling, processing and validation.Int J Biol Sci. 2018 Sep 7;14(12):1709-1714. doi: 10.7150/ijbs.27168. eCollection 2018. Int J Biol Sci. 2018. PMID: 30416385 Free PMC article. Review.
References
-
- Organization, W. H. Coronavirus disease (COVID-19) pandemic 2024.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
