

Functional analysis of a novel FOXL2 mutation in blepharophimosis, ptosis, and epicanthus inversus syndrome type II and elucidation of the genotype-phenotype correlation
- PMID: 40251640
- PMCID: PMC12008864
- DOI: 10.1186/s40246-025-00752-7
Functional analysis of a novel FOXL2 mutation in blepharophimosis, ptosis, and epicanthus inversus syndrome type II and elucidation of the genotype-phenotype correlation
Abstract
Background: Blepharophimosis, ptosis, and epicanthus inversus syndrome (BPES) is a rare autosomal dominant disorder caused by genetic mutations. However, the genotype-phenotype correlation remains unclear. This study aimed to identify mutations in a Chinese family with BPES and elucidate the genotype-phenotype relationship.
Methods: A comprehensive clinical and molecular genetic analysis was conducted on a three-generation Chinese family with BPES, which was prospectively enrolled at the Eye Hospital of Wenzhou Medical University. Affected individuals underwent systematic phenotyping, including detailed physical and ophthalmic evaluations. Genomic DNA was isolated from peripheral blood samples and subjected to whole-exome sequencing, followed by targeted Sanger sequencing for variant validation. Candidate disease-associated variants were analyzed using in silico predictive algorithms to assess their potential structural and functional impact on encoded proteins. To further elucidate the pathogenicity of the identified mutation, functional studies were performed, including immunofluorescence-based subcellular localization assays and quantitative real-time PCR to evaluate transcriptional regulatory effects.
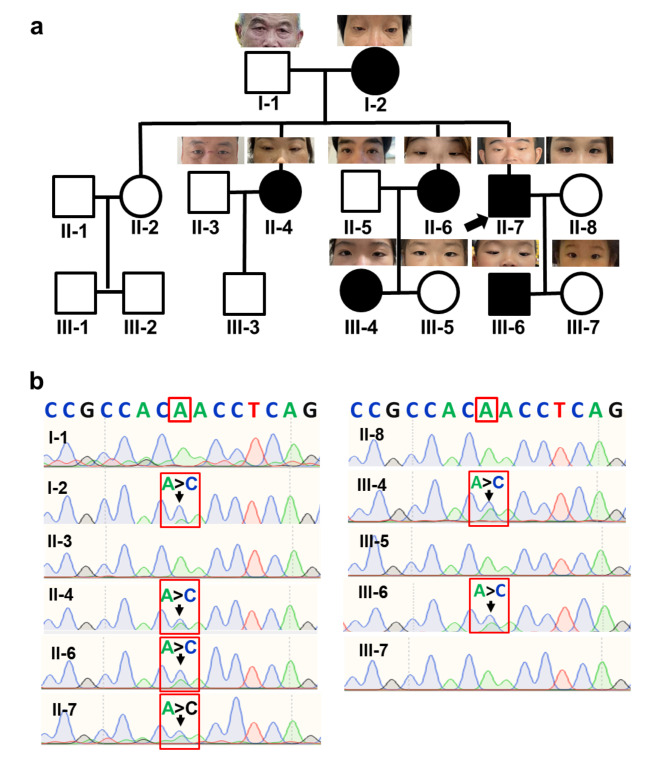
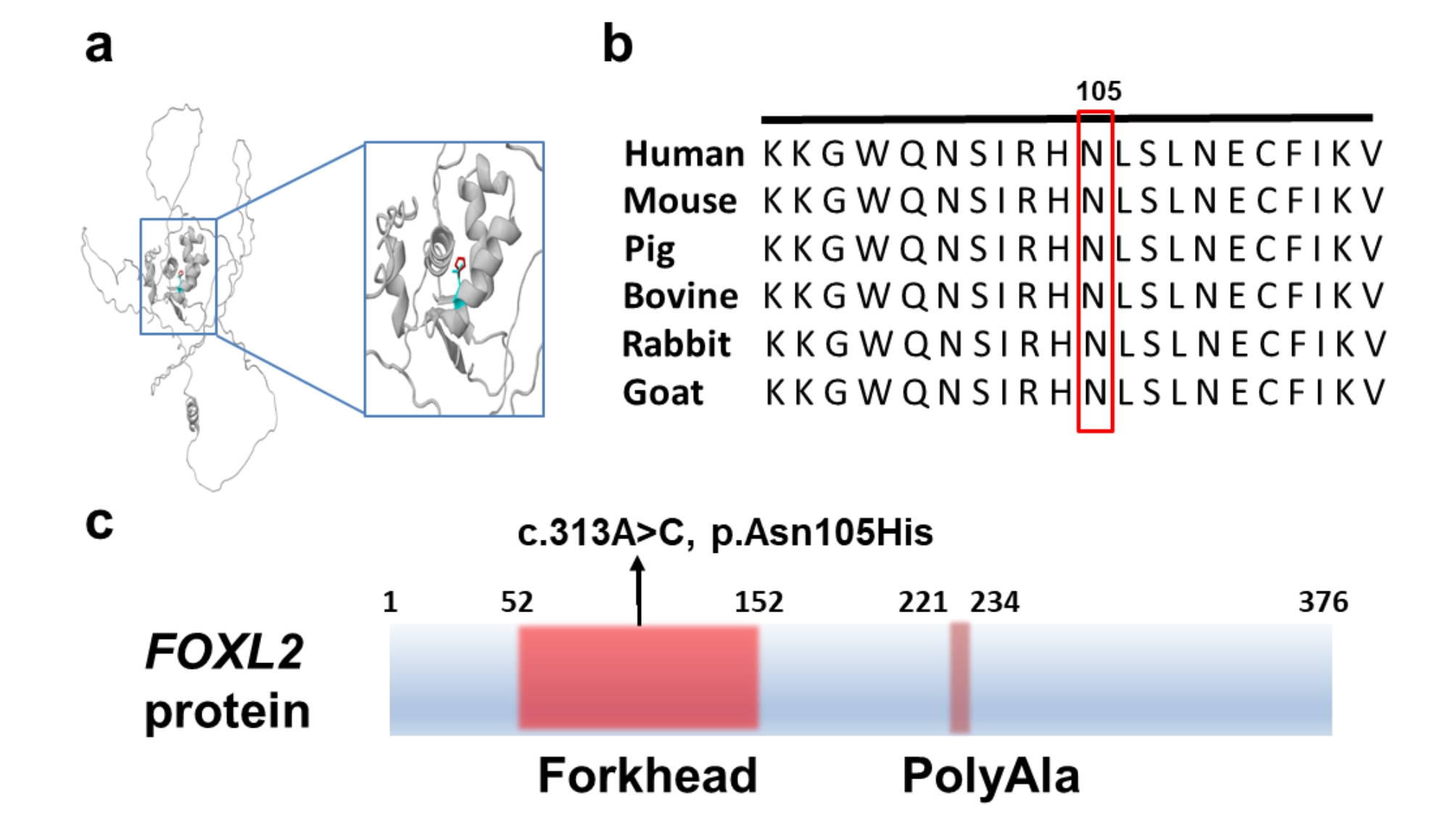
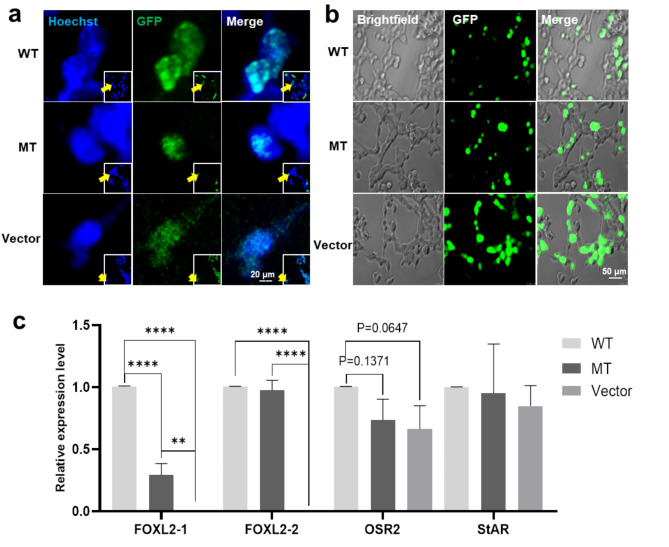
Results: Six affected individuals of this pedigree presented with canonical BPES features including small palpebral fissures, ptosis, epicanthus inversus, and telecanthus, without premature ovarian failure, consistent with a diagnosis of BPES type II. Whole-exome sequencing revealed a heterozygous missense mutation (c.313 A > C:p.N105H) in FOXL2, which was subsequently validated by Sanger sequencing. This variant demonstrated complete cosegregation with the BPES phenotype across all affected family members. According to ACMG guidelines, the variant was classified as Likely Pathogenic (PS1 + PM1 + PM2 + PP3). In silico pathogenicity prediction tools classified the p.N105H variant as deleterious. Immunofluorescence assays revealed aberrant nuclear aggregation of the mutant FOXL2 protein, and functional characterization via quantitative real-time PCR demonstrated no significant dysregulation (P > 0.05) of downstream targets (STAR, OSR2).
Conclusions: This study provides functional evidence of the pathogenic FOXL2 mutation (c.313 A > C, p.N105H) in BPES type II, demonstrating its disruptive effects on protein localization while maintaining normal transcriptional activity of downstream targets. These findings expand the mutational spectrum of FOXL2 related disorders and enhance our understanding of genotype-phenotype correlations in BPES.
Keywords: Blepharophimosis-ptosis-epicanthus inversus syndrome; FOXL2; Genotype‒phenotype correlation; Pathogenic variants; Whole-exome sequencing.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the Ethics Committee of the Eye Hospital of Wenzhou Medical University (Date: 2022-04-07, No: 2022-046-K-31-01). Written informed consent was obtained from all individual participants or their legal guardians (for participants under 16 years of age) included in the study. Consent for publication: The authors affirm that human research participants provided informed consent for publication of the images in Fig. 1a. Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
Functional Analysis of a Novel FOXL2 Indel Mutation in Chinese Families with Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome Type I.Int J Biol Sci. 2017 Jul 18;13(8):1019-1028. doi: 10.7150/ijbs.19532. eCollection 2017. Int J Biol Sci. 2017. PMID: 28924383 Free PMC article.
-
Identification of a novel FOXL2 mutation in a single family with both types of blepharophimosis‑-ptosis-epicanthus inversus syndrome.Mol Med Rep. 2017 Oct;16(4):5529-5532. doi: 10.3892/mmr.2017.7226. Epub 2017 Aug 10. Mol Med Rep. 2017. PMID: 28849110
-
Genetic and Functional Analyses of Two Missense Mutations in the Transcription Factor FOXL2 in Two Chinese Families with Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome.Genet Test Mol Biomarkers. 2018 Oct;22(10):585-592. doi: 10.1089/gtmb.2018.0064. Epub 2018 Sep 20. Genet Test Mol Biomarkers. 2018. PMID: 30234390
-
The Genetic and Clinical Features of FOXL2-Related Blepharophimosis, Ptosis and Epicanthus Inversus Syndrome.Genes (Basel). 2021 Mar 4;12(3):364. doi: 10.3390/genes12030364. Genes (Basel). 2021. PMID: 33806295 Free PMC article. Review.
-
Blepharophimosis, Ptosis, and Epicanthus Inversus Syndrome.2004 Jul 8 [updated 2022 Mar 10]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2004 Jul 8 [updated 2022 Mar 10]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 20301614 Free Books & Documents. Review.
References
-
- Verdin H, Matton C, De Baere E, Blepharophimosis et al. Ptosis, and Epicanthus Inversus Syndrome. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, editors. GeneReviews((R)). Seattle (WA)1993. - PubMed
-
- De Vos M, Devroey P, Fauser BC. Primary ovarian insufficiency. Lancet. 2010;376(9744):911–21. 10.1016/S0140-6736(10)60355-8 - PubMed
-
- Stromme P, Sandboe F. Blepharophimosis-ptosis-epicanthus inversus syndrome (BPES). Acta Ophthalmol Scand. 1996;74(1):45–7. 10.1111/j.1600-0420.1996.tb00680.x - PubMed
-
- Beysen D, De Paepe A, De Baere E. FOXL2 mutations and genomic rearrangements in BPES. Hum Mutat. 2009;30(2):158–69. 10.1002/humu.20807 - PubMed
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
