

16p13.11 microduplication with growth retardation and developmental disorders: a case report and literature review
- PMID: 40256007
- PMCID: PMC12003995
- DOI: 10.18999/nagjms.87.1.144
16p13.11 microduplication with growth retardation and developmental disorders: a case report and literature review
Abstract
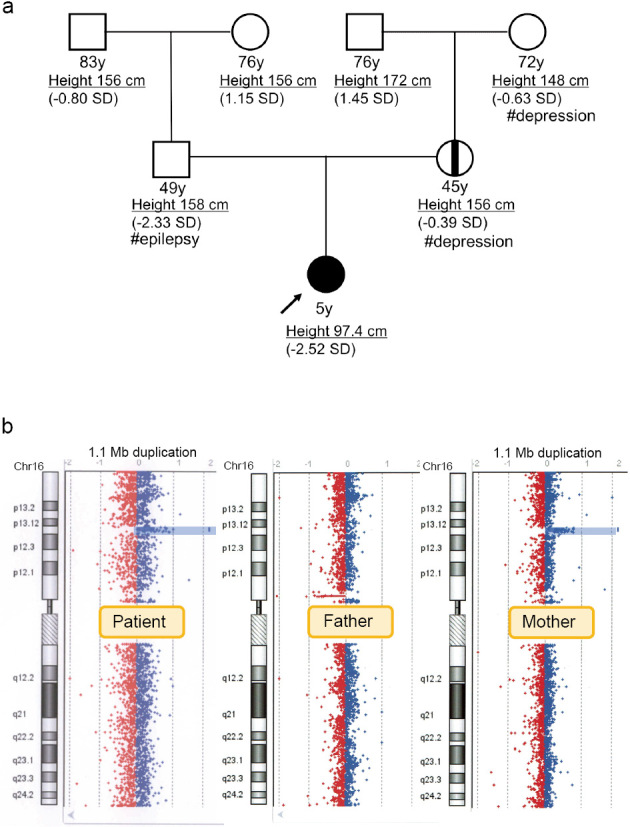
Short stature and growth retardation is a common condition in children. Genetic variations are responsible for many cases of short stature of unknown etiology. In particular, pathogenic copy number variants (CNVs) have been found in 10%-16% of children with unexplained short stature. This paper reports on a 5-year-old Japanese girl with both growth retardation and developmental delay associated with a 16p13.11 microduplication. Although the patient's mother also carries this microduplication, she did not show growth retardation and developmental delay. These cases illustrate the diverse phenotypic manifestations of 16p13.11 microduplication. Consequently, we conducted the literature review of 274 cases associated with this duplication revealed neurological disorders in approximately 70% of cases, 15.3% of these cases were associated with short stature. Diagnosis of 16p13.11 microduplication remains challenging due to its diverse symptomatology and elusive genotype-phenotype correlations. Comprehensive genetic evaluation is crucial for patients presenting with short stature and developmental disorders, underscoring the need for further investigation into the 16p13.11 microduplication to clarify its specific role and implications.
Keywords: 16p13.11 microduplication; copy number variants (CNVs); developmental disorders; growth retardation.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Concurrent de novo MACF1 mutation and inherited 16p13.11 microduplication in a preterm newborn with hypotonia, joint hyperlaxity and multiple congenital malformations: a case report.BMC Pediatr. 2024 Aug 16;24(1):528. doi: 10.1186/s12887-024-04628-y. BMC Pediatr. 2024. PMID: 39152427 Free PMC article.
-
Male-biased autosomal effect of 16p13.11 copy number variation in neurodevelopmental disorders.PLoS One. 2013 Apr 18;8(4):e61365. doi: 10.1371/journal.pone.0061365. Print 2013. PLoS One. 2013. PMID: 23637818 Free PMC article.
-
A maternally inherited 16p13.11-p12.3 duplication concomitant with a de novo SOX5 deletion in a male patient with global developmental delay, disruptive and obsessive behaviors and minor dysmorphic features.Am J Med Genet A. 2015 Jun;167(6):1315-22. doi: 10.1002/ajmg.a.36909. Epub 2015 Apr 2. Am J Med Genet A. 2015. PMID: 25847113
-
Phenotypic expansion of the interstitial 16p13.3 duplication: a case report and review of the literature.Gene. 2013 Dec 1;531(2):502-5. doi: 10.1016/j.gene.2013.09.006. Epub 2013 Sep 12. Gene. 2013. PMID: 24035902 Review.
-
Expanding the clinical spectrum of 19p13.3 microduplication syndrome: a case report highlighting nephrotic syndrome and literature review.BMC Pediatr. 2025 Jan 28;25(1):70. doi: 10.1186/s12887-025-05394-1. BMC Pediatr. 2025. PMID: 39875952 Free PMC article. Review.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical