

A novel compound heterozygous mutation (c.64G > A and c.506-1G > A) associated with congenital coagulation factor VII deficiency: a case report and literature review
- PMID: 40257480
- PMCID: PMC12141372
- DOI: 10.1007/s00277-025-06364-4
A novel compound heterozygous mutation (c.64G > A and c.506-1G > A) associated with congenital coagulation factor VII deficiency: a case report and literature review
Abstract
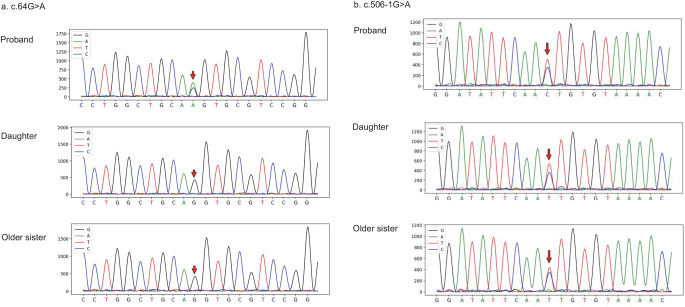
Congenital factor VII (FVII) deficiency is a rare autosomal recessive bleeding disorder characterized by prolonged prothrombin time (PT) and reduced FVII coagulant activity (FVII: C). Here, we present the case of a middle-aged male patient with gastrointestinal bleeding, who exhibited prolonged PT and decreased FVII: C levels. Gene sequencing analysis revealed compound heterozygous mutations in the F7 gene: c.64G > A (p.V22I) and c.506-1G > A. Based on the laboratory results and gene sequencing, the patient was diagnosed as FVII deficiency. After adding recombinant activated FVII (rFVIIa) for several days, the laboratory indicators returned to normal and the bleeding symptoms were relieved. In subsequent validation studies, we also identified the c.506-1G > A mutation in his older sister and daughter. Importantly, this represents the first documented case where both mutations coexist concurrently. Additionally, our literature review reveals that approximately 50% of mutation types associated with congenital FVII deficiency are located on exon 9; however, there is no significant correlation between the reduction in FVII: C levels and severity of clinical symptoms based on EAHAD database analysis.
Keywords: Case report; Congenital factor VII deficiency; EAHAD; Prothrombin time.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethical standards and consent to publish declaration: The studies involving participant was reviewed and approved by the Ethics Committee of The First Hospital of China Medical University. The patient and his family provided their written informed consent to take part in this study. A copy of the written consent is available for review by the editorial office of this journal. Competing interests: The authors declare no competing interests.
Figures


Similar articles
-
A novel missense mutation close to the charge-stabilizing system in a patient with congenital factor VII deficiency.Blood Coagul Fibrinolysis. 2011 Jun;22(4):264-70. doi: 10.1097/MBC.0b013e3283447388. Blood Coagul Fibrinolysis. 2011. PMID: 21372693
-
Novel heterozygous F7 gene mutation (c. C1286T) associated with congenital factor VII deficiency: A case report and literature review.J Clin Lab Anal. 2022 May;36(5):e24349. doi: 10.1002/jcla.24349. Epub 2022 Mar 29. J Clin Lab Anal. 2022. PMID: 35349734 Free PMC article. Review.
-
F7 gene variants modulate protein levels in a large cohort of patients with factor VII deficiency. Results from a genotype-phenotype study.Thromb Haemost. 2017 Aug 1;117(8):1455-1464. doi: 10.1160/TH17-02-0085. Epub 2017 Apr 27. Thromb Haemost. 2017. PMID: 28447100
-
Identification of two novel mutations in three children with congenital factor VII deficiency.Blood Coagul Fibrinolysis. 2021 Jul 1;32(5):340-343. doi: 10.1097/MBC.0000000000001022. Blood Coagul Fibrinolysis. 2021. PMID: 33587484 Free PMC article.
-
Factor VII deficiency: defining the clinical picture and optimizing therapeutic options.Haemophilia. 2008 Nov;14(6):1170-5. doi: 10.1111/j.1365-2516.2008.01844.x. Haemophilia. 2008. PMID: 19141157 Review.
References
-
- Palla R, Peyvandi F, Shapiro AD (2015) Rare bleeding disorders: diagnosis and treatment [J]. Blood 125(13):2052–2061 - PubMed
-
- Sevenet PO, Kaczor DA, Depasse F, Factor VII, Deficiency (2017) From basics to clinical laboratory diagnosis and patient management [J]. Clin Appl Thromb Hemost 23(7):703–710 - PubMed
-
- Mariani G, Bernardi F, Factor VII, Deficiency (2009) [J] Semin Thromb Hemost 35(4):400–406 - PubMed
-
- Giansily-Blaizot M, Rallapalli PM, Perkins SJ et al (2020) The EAHAD blood coagulation factor VII variant database [J]. Hum Mutat 41(7):1209–1219 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
