

Revealing cancer driver genes through integrative transcriptomic and epigenomic analyses with Moonlight
- PMID: 40258059
- PMCID: PMC12058160
- DOI: 10.1371/journal.pcbi.1012999
Revealing cancer driver genes through integrative transcriptomic and epigenomic analyses with Moonlight
Abstract
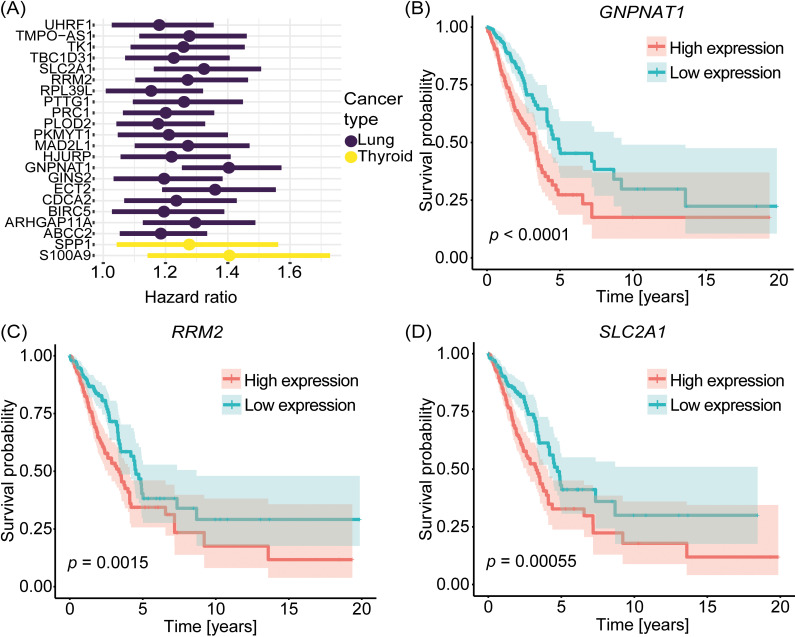
Cancer involves dynamic changes caused by (epi)genetic alterations such as mutations or abnormal DNA methylation patterns which occur in cancer driver genes. These driver genes are divided into oncogenes and tumor suppressors depending on their function and mechanism of action. Discovering driver genes in different cancer (sub)types is important not only for increasing current understanding of carcinogenesis but also from prognostic and therapeutic perspectives. We have previously developed a framework called Moonlight which uses a systems biology multi-omics approach for prediction of driver genes. Here, we present an important development in Moonlight2 by incorporating a DNA methylation layer which provides epigenetic evidence for deregulated expression profiles of driver genes. To this end, we present a novel functionality called Gene Methylation Analysis (GMA) which investigates abnormal DNA methylation patterns to predict driver genes. This is achieved by integrating the tool EpiMix which is designed to detect such aberrant DNA methylation patterns in a cohort of patients and further couples these patterns with gene expression changes. To showcase GMA, we applied it to three cancer (sub)types (basal-like breast cancer, lung adenocarcinoma, and thyroid carcinoma) where we discovered 33, 190, and 263 epigenetically driven genes, respectively. A subset of these driver genes had prognostic effects with expression levels significantly affecting survival of the patients. Moreover, a subset of the driver genes demonstrated therapeutic potential as drug targets. This study provides a framework for exploring the driving forces behind cancer and provides novel insights into the landscape of three cancer sub(types) by integrating gene expression and methylation data.
Copyright: © 2025 Nourbakhsh et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures






Similar articles
-
An integrated epigenomic-transcriptomic landscape of lung cancer reveals novel methylation driver genes of diagnostic and therapeutic relevance.Theranostics. 2021 Mar 11;11(11):5346-5364. doi: 10.7150/thno.58385. eCollection 2021. Theranostics. 2021. PMID: 33859751 Free PMC article.
-
Identification of candidate cancer drivers by integrative Epi-DNA and Gene Expression (iEDGE) data analysis.Sci Rep. 2019 Nov 15;9(1):16904. doi: 10.1038/s41598-019-52886-z. Sci Rep. 2019. PMID: 31729402 Free PMC article.
-
Protocol for Analyzing Epigenetic Regulation Mechanisms in Breast Cancer.Methods Mol Biol. 2024;2812:275-306. doi: 10.1007/978-1-0716-3886-6_16. Methods Mol Biol. 2024. PMID: 39068369
-
Epigenome remodelling in breast cancer: insights from an early in vitro model of carcinogenesis.Breast Cancer Res. 2012 Nov 15;14(6):215. doi: 10.1186/bcr3237. Breast Cancer Res. 2012. PMID: 23168266 Free PMC article. Review.
-
DNA methylation aberrancies delineate clinically distinct subsets of colorectal cancer and provide novel targets for epigenetic therapies.Oncogene. 2018 Feb 1;37(5):566-577. doi: 10.1038/onc.2017.374. Epub 2017 Oct 9. Oncogene. 2018. PMID: 28991233 Free PMC article. Review.
References
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
