

rhCC16 Suppresses Cellular Senescence and Ameliorates COPD-Like Symptoms by Activating the AMPK/Sirt1-PGC-1-α-TFAM Pathway to Promote Mitochondrial Function
- PMID: 40259209
- PMCID: PMC12011551
- DOI: 10.1111/jcmm.70566
rhCC16 Suppresses Cellular Senescence and Ameliorates COPD-Like Symptoms by Activating the AMPK/Sirt1-PGC-1-α-TFAM Pathway to Promote Mitochondrial Function
Abstract
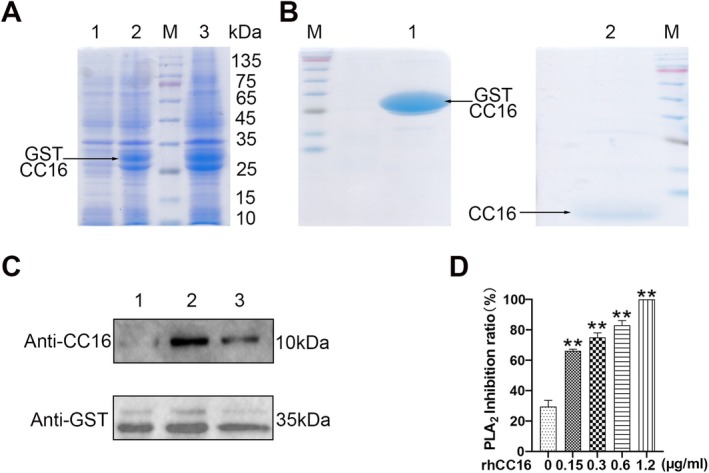
Chronic obstructive pulmonary disease (COPD) is a widespread lung disease marked by alveolar wall damage, leading to inflammation and fibrosis. Key risk factors include age, smoking, sex, and education, with smoking being the most crucial. These factors are globally consistent and linked with aging. Club cell secretory protein 16 (CC16), primarily secreted by non-ciliated bronchial epithelial cells, is crucial for pulmonary health, offering anti-inflammatory and antioxidant benefits. CC16 levels are notably reduced in COPD, suggesting its enhancement as a potential treatment. In this study, cellular senescence of BEAS-2B cells was stimulated using cigarette smoke extract (CSE) and the function of recombinant human CC16 protein (rhCC16) in cellular senescence was assessed by detecting the levels of β-galactosidase, p16, p21, ROS and the underlined mechanism was revealed by measuring mitochondrial biogenesis and metabolism. Additionally, COPD mice were prepared, and rhCC16's role on the cellular senescence of lung tissues was examined. Our findings showed that rhCC16 ameliorated cellular senescence in BEAS-2B cells and lung tissues of COPD mice accompanied by lower levels of β-galactosidase, p16, p21 and ROS. Mechanically, rhCC16 mitigated senescence via triggering PGC-1α expression through the AMPK/SIRT1 pathway and fostering mitochondrial biogenesis and metabolism to reduce the levels of ROS. Furthermore, the results also indicated that rhCC16 exerted its effect via both integrin α4β1 and clathrin-mediated endocytosis. Collectively, rhCC16 suppresses cellular senescence and ameliorates COPD-like symptoms by activating the AMPK/Sirt1-PGC-1-α-TFAM pathway to foster mitochondrial function.
Keywords: AMPK; COPD; cellular senescence; mitochondrial function; rhCC16.
© 2025 The Author(s). Journal of Cellular and Molecular Medicine published by Foundation for Cellular and Molecular Medicine and John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures





Similar articles
-
[Shenqi Buzhong Formula ameliorates mitochondrial dysfunction in a rat model of chronic obstructive pulmonary disease by activating the AMPK/SIRT1/PGC-1α pathway].Nan Fang Yi Ke Da Xue Xue Bao. 2025 May 20;45(5):969-976. doi: 10.12122/j.issn.1673-4254.2025.05.09. Nan Fang Yi Ke Da Xue Xue Bao. 2025. PMID: 40415428 Free PMC article. Chinese.
-
Progesterone (P4) ameliorates cigarette smoke-induced chronic obstructive pulmonary disease (COPD).Mol Med. 2024 Aug 13;30(1):123. doi: 10.1186/s10020-024-00883-y. Mol Med. 2024. PMID: 39138434 Free PMC article.
-
The diabetes medication canagliflozin promotes mitochondrial remodelling of adipocyte via the AMPK-Sirt1-Pgc-1α signalling pathway.Adipocyte. 2020 Dec;9(1):484-494. doi: 10.1080/21623945.2020.1807850. Adipocyte. 2020. PMID: 32835596 Free PMC article.
-
Role of mitochondria in diabetic peripheral neuropathy: Influencing the NAD+-dependent SIRT1-PGC-1α-TFAM pathway.Int Rev Neurobiol. 2019;145:177-209. doi: 10.1016/bs.irn.2019.04.002. Epub 2019 Jun 8. Int Rev Neurobiol. 2019. PMID: 31208524 Free PMC article. Review.
-
Molecular pathogenesis in chronic obstructive pulmonary disease and therapeutic potential by targeting AMP-activated protein kinase.J Cell Physiol. 2018 Mar;233(3):1999-2006. doi: 10.1002/jcp.25844. Epub 2017 Apr 27. J Cell Physiol. 2018. PMID: 28160496 Review.
References
-
- Mannino D. M. and Buist A. S., “Global Burden of COPD: Risk Factors, Prevalence, and Future Trends,” Lancet 370 (2007): 765–773. - PubMed
-
- Rutten E. P. A., Gopal P., Wouters E. F. M., et al., “Various Mechanistic Pathways Representing the Aging Process Are Altered in COPD,” Chest 149 (2016): 53–61. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
