

Custom CRISPR-Cas9 PAM variants via scalable engineering and machine learning
- PMID: 40262634
- PMCID: PMC12449813
- DOI: 10.1038/s41586-025-09021-y
Custom CRISPR-Cas9 PAM variants via scalable engineering and machine learning
Abstract
Engineering and characterizing proteins can be time-consuming and cumbersome, motivating the development of generalist CRISPR-Cas enzymes1-4 to enable diverse genome-editing applications. However, such enzymes have caveats such as an increased risk of off-target editing3,5,6. Here, to enable scalable reprogramming of Cas9 enzymes, we combined high-throughput protein engineering with machine learning to derive bespoke editors that are more uniquely suited to specific targets. Through structure-function-informed saturation mutagenesis and bacterial selections, we obtained nearly 1,000 engineered SpCas9 enzymes and characterized their protospacer-adjacent motif (PAM)7 requirements to train a neural network that relates amino acid sequence to PAM specificity. By utilizing the resulting PAM machine learning algorithm (PAMmla) to predict the PAMs of 64 million SpCas9 enzymes, we identified efficacious and specific enzymes that outperform evolution-based and engineered SpCas9 enzymes as nucleases and base editors in human cells while reducing off-targets. An in silico-directed evolution method enables user-directed Cas9 enzyme design, including for allele-selective targeting of the RHOP23H allele in human cells and mice. Together, PAMmla integrates machine learning and protein engineering to curate a catalogue of SpCas9 enzymes with distinct PAM requirements, motivating a shift away from generalist enzymes towards safe and efficient bespoke Cas9 variants.
© 2025. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Competing interests: R.A.S. and B.P.K. are inventors on a patent application filed by Mass General Brigham (MGB) that describes the development of PAMmla. B.P.K. and R.T.W. are inventors on additional patents or patent applications filed by MGB that describe genome engineering technologies related to the current study. S.Q.T. is an inventor on a patent application for GUIDE-seq, and is a member of the scientific advisory boards of Ensoma and Prime Medicine. L.P. has financial interests in Edilytics and SeQure Dx. Q.L. is a consultant for Entrada Therapeutics. B.P.K. is a consultant for EcoR1 capital, Novartis Venture Fund and Jumble Therapeutics, and is on the scientific advisory boards of Acrigen Biosciences, Life Edit Therapeutics and Prime Medicine. B.P.K. has a financial interest in Prime Medicine, Inc., a company developing therapeutic CRISPR–Cas technologies for gene editing. The interests of L.P. and B.P.K. were reviewed and are managed by MGH and MGB in accordance with their conflict-of-interest policies. The other authors declare no competing interests.
Figures




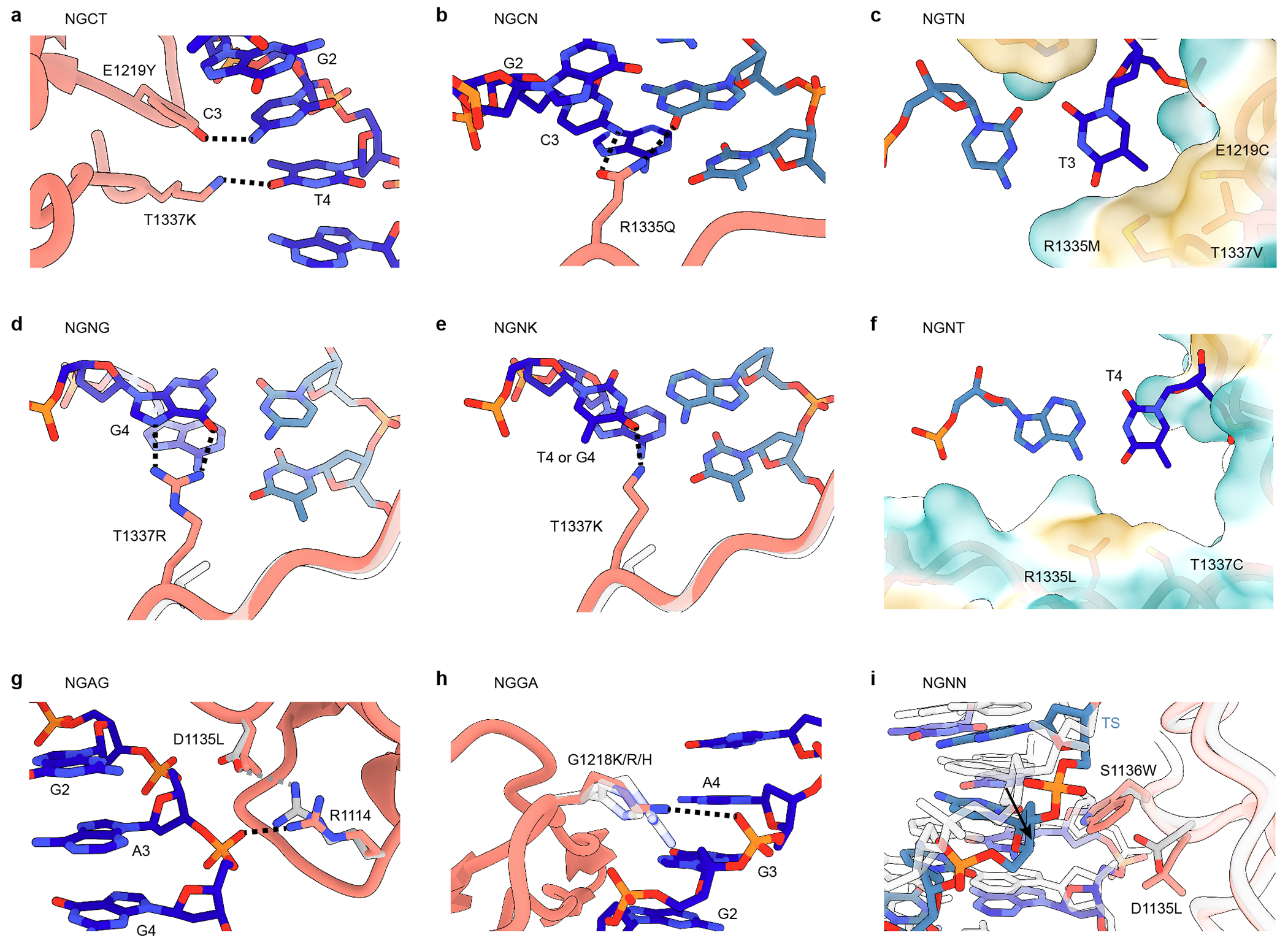
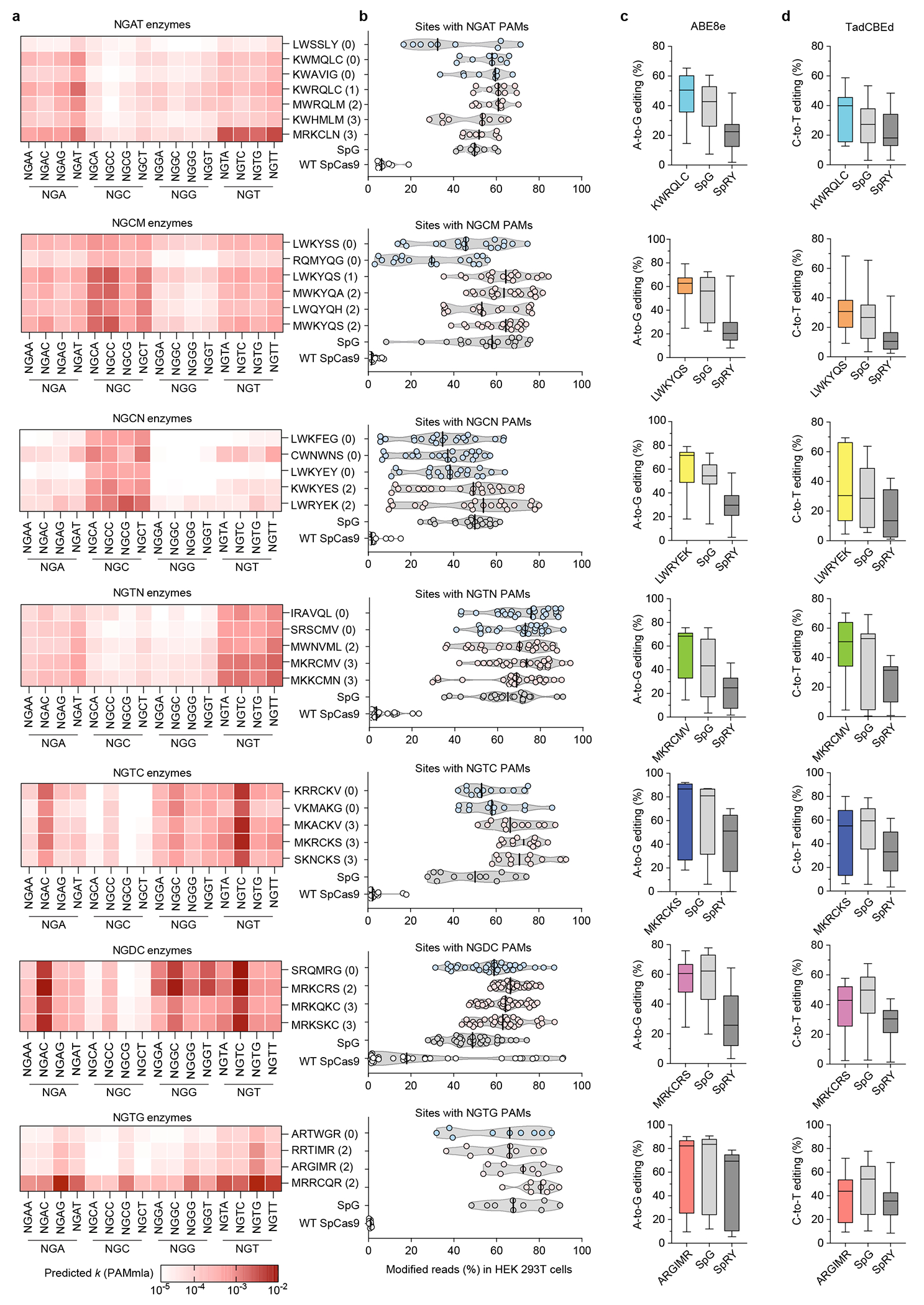






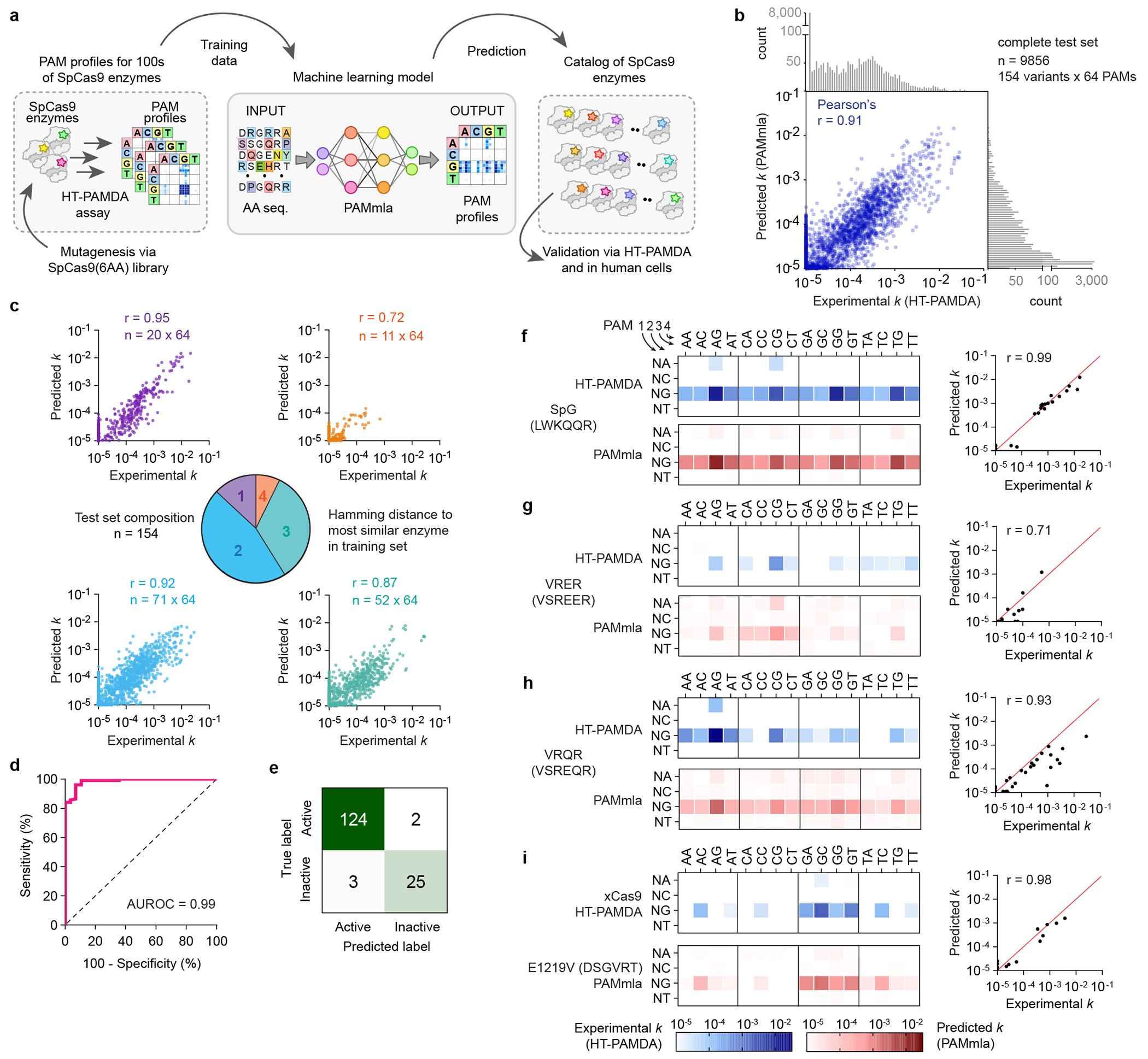



References
METHODS SECTION-ONLY REFERENCES
-
- Gibson DG et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods 2009 6:5 6, 343–345 (2009). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
