

HPOseq: a deep ensemble model for predicting the protein-phenotype relationships based on protein sequences
- PMID: 40263997
- PMCID: PMC12013097
- DOI: 10.1186/s12859-025-06122-3
HPOseq: a deep ensemble model for predicting the protein-phenotype relationships based on protein sequences
Abstract
Background: Understanding the relationships between proteins and specific disease phenotypes contributes to the early detection of diseases and advances the development of personalized medicine. The acquisition of a large amount of proteomics data has facilitated this process. To improve discovery efficiency and reduce the time and financial costs associated with biological experiments, various computational methods have yielded promising results. However, the lack of rich and reliable protein-related information still presents challenges in this process.
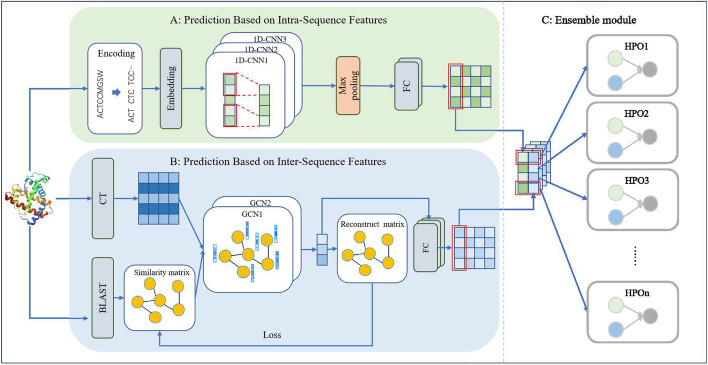
Results: In this paper, we propose an ensemble prediction model, named HPOseq, which predicts human protein-phenotype relationships based only on sequence information. HPOseq establishes two base models to achieve objectives. One directly extracts internal information from amino acid sequences as protein features to predict the associated phenotypes. The other builds a protein-protein network based on sequence similarity, extracting information between proteins for phenotype prediction. Ultimately, an ensemble module is employed to integrate the predictions from both base models, resulting in the final prediction.
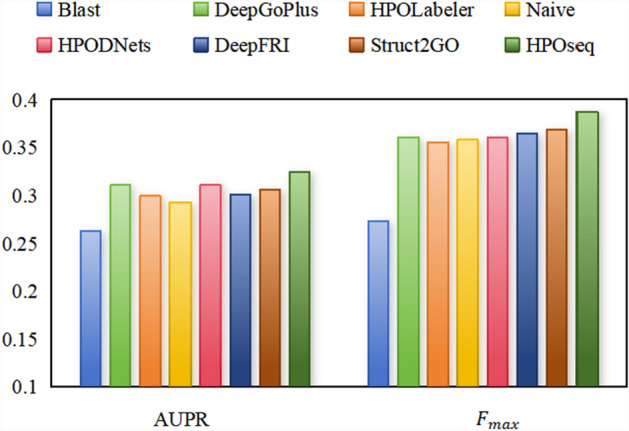
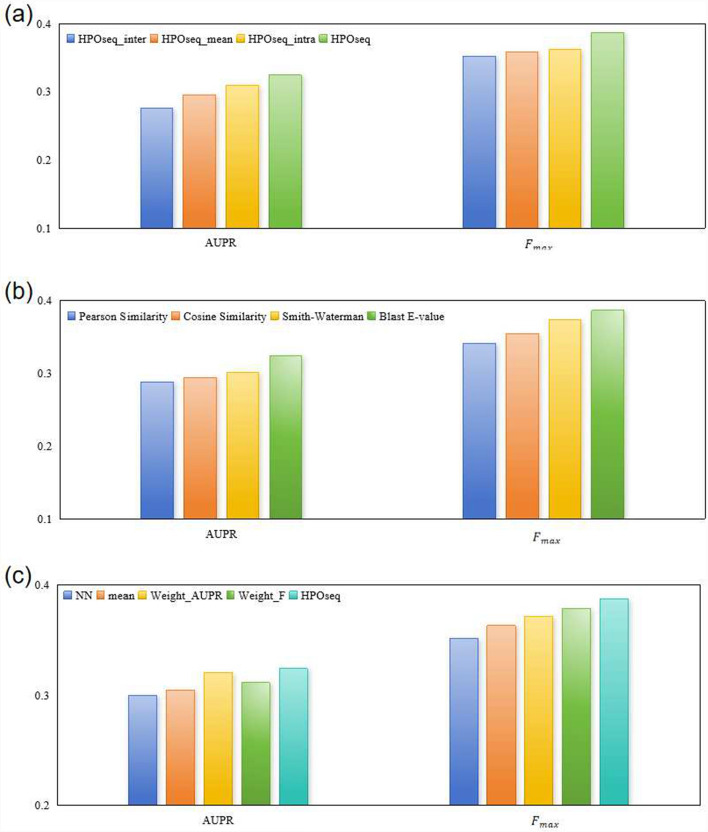
Conclusion: The results of 5-fold cross-validation reveal that HPOseq outperforms seven baseline methods for predicting protein-phenotype relationships. Moreover, we conduct case studies from the points of phenotype annotation and protein analysis to verify the practical significance of HPOseq.
Keywords: Amino acid sequence; Deep learning; Ensemble model; Variational graph autoencoder.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Not applicable. Conflict of interest: The authors declare that they have no Conflict of interest.
Figures

References
-
- Bao W, Yang B. Protein acetylation sites with complex-valued polynomial model. Front Comput Sci. 2024;18: 183904.
-
- Bao W, Chen B, Zhang Y. WSHNN: A weakly supervised hybrid neural network for the identification of DNA-protein binding sites. Curr Comput-aided Drug Design. 2024. 10.2174/0115734099277249240129114123. - PubMed
MeSH terms
Substances
Grants and funding
- 2022B03023/the Key R&D Program of Xinjiang Uygur Autonomous Regin
- 2022B03023/the Key R&D Program of Xinjiang Uygur Autonomous Regin
- 2022B03023/the Key R&D Program of Xinjiang Uygur Autonomous Regin
- 2022B03023/the Key R&D Program of Xinjiang Uygur Autonomous Regin
- 2022B03023/the Key R&D Program of Xinjiang Uygur Autonomous Regin
- 2022B03023/the Key R&D Program of Xinjiang Uygur Autonomous Regin
- 2022B03023/the Key R&D Program of Xinjiang Uygur Autonomous Regin
- 2024D01C126/the Natural Science Foundation of Xinjiang Uygur Autonomous Regin
- 2024D01C126/the Natural Science Foundation of Xinjiang Uygur Autonomous Regin
- 2024D01C126/the Natural Science Foundation of Xinjiang Uygur Autonomous Regin
- 2024D01C126/the Natural Science Foundation of Xinjiang Uygur Autonomous Regin
- 2024D01C126/the Natural Science Foundation of Xinjiang Uygur Autonomous Regin
- 2024D01C126/the Natural Science Foundation of Xinjiang Uygur Autonomous Regin
- 2024D01C126/the Natural Science Foundation of Xinjiang Uygur Autonomous Regin
- 62366052/the Natural Science Foundation of China
- 62366052/the Natural Science Foundation of China
- 62366052/the Natural Science Foundation of China
- 62366052/the Natural Science Foundation of China
- 62366052/the Natural Science Foundation of China
- 62366052/the Natural Science Foundation of China
- 62366052/the Natural Science Foundation of China
LinkOut - more resources
Full Text Sources
