

Mitochondrial DNA signals driving immune responses: Why, How, Where?
- PMID: 40264103
- PMCID: PMC12012978
- DOI: 10.1186/s12964-025-02042-0
Mitochondrial DNA signals driving immune responses: Why, How, Where?
Abstract
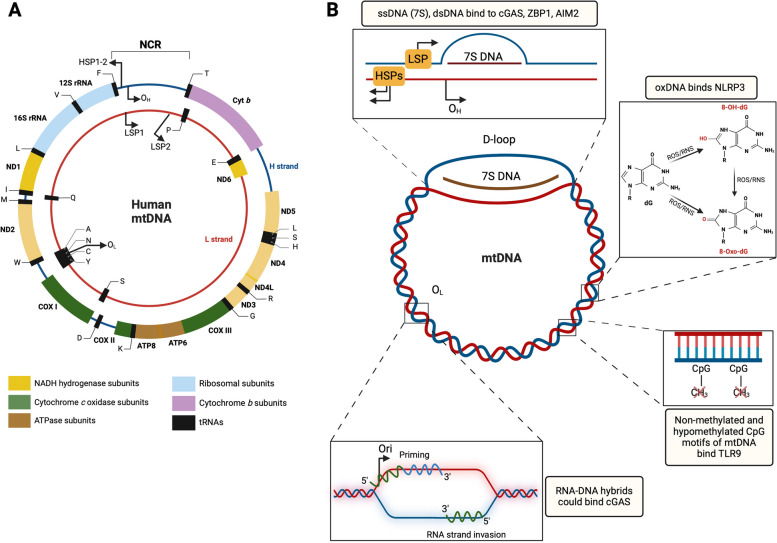
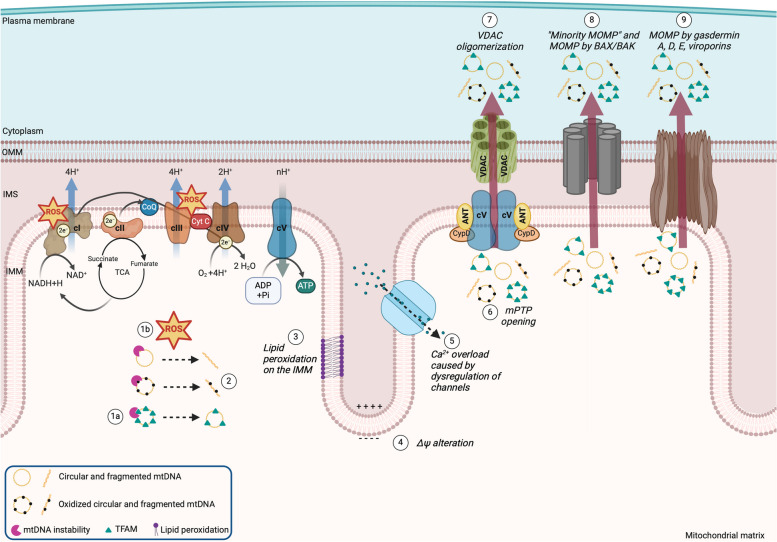
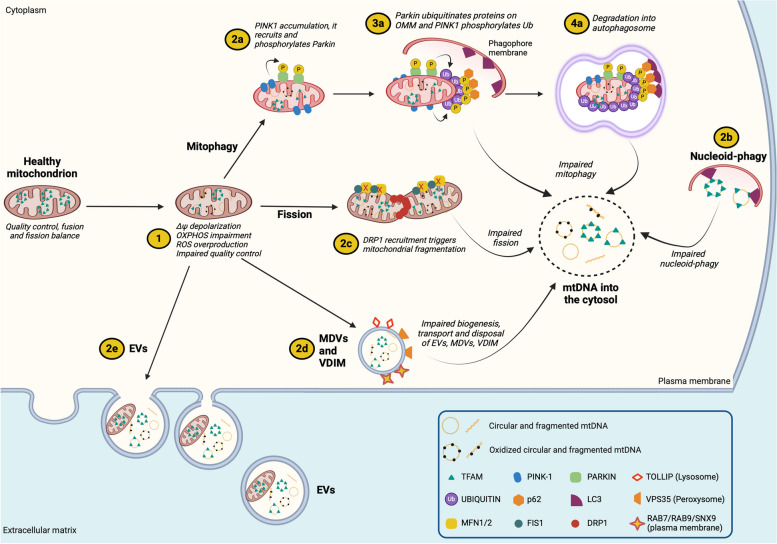
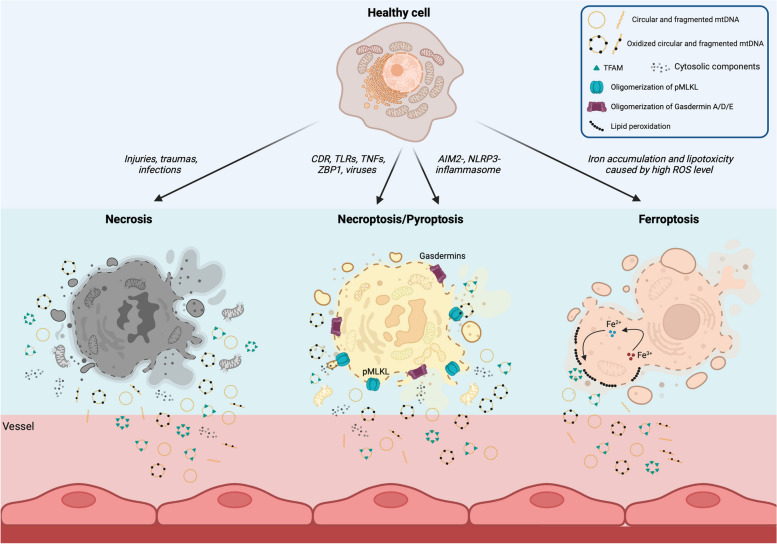
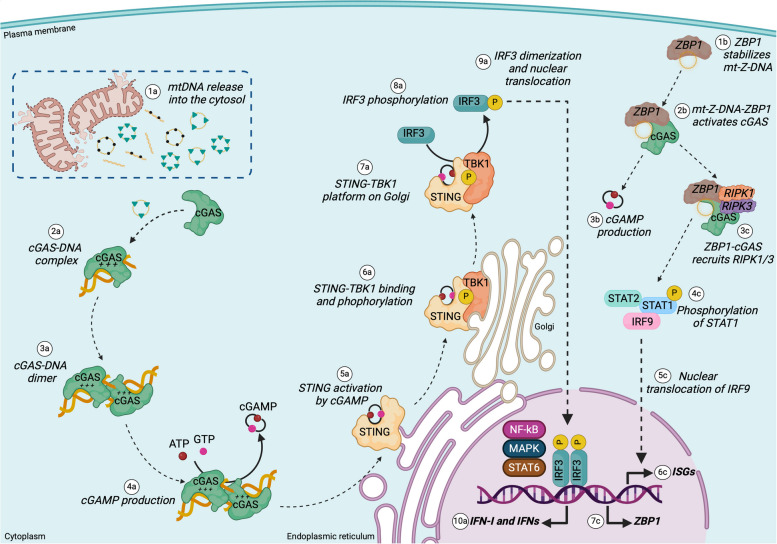
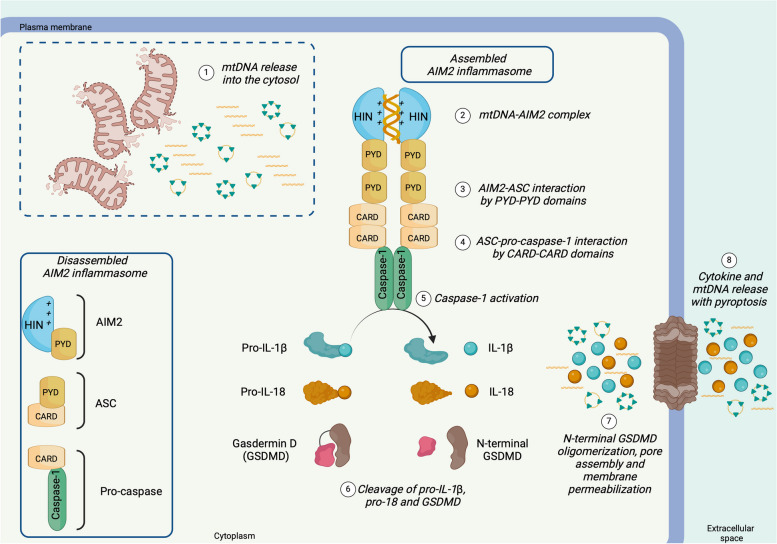
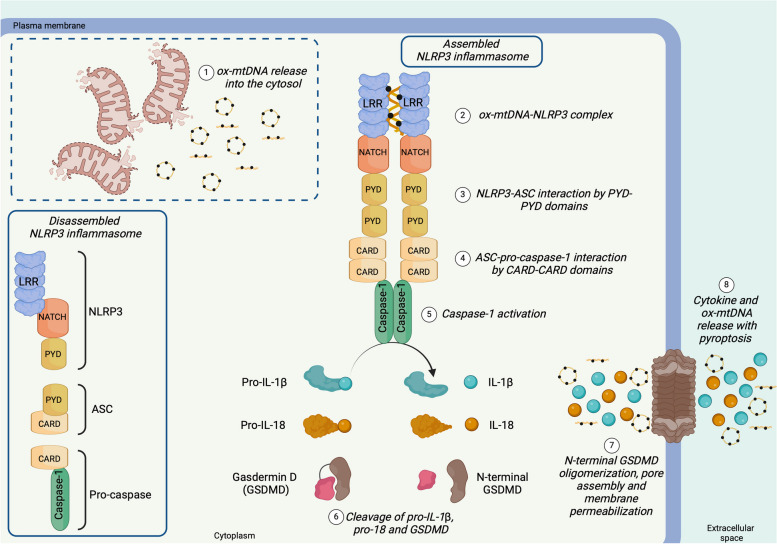
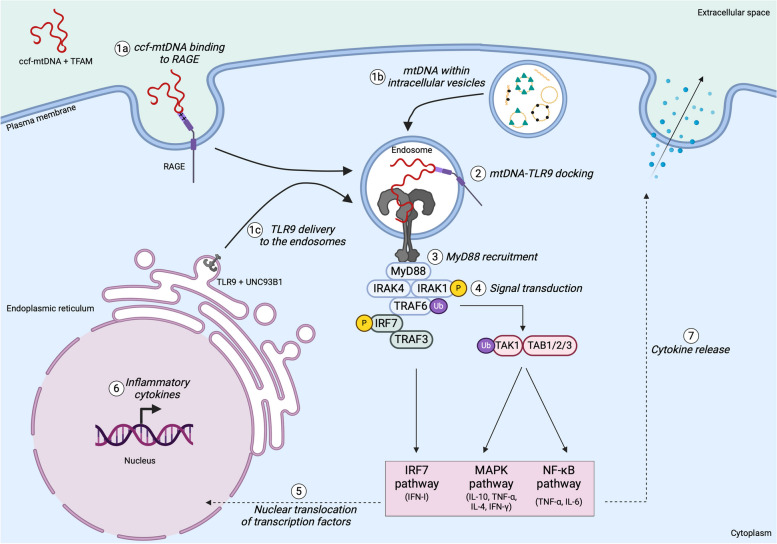
There has been a recent expansion in our understanding of DNA-sensing mechanisms. Mitochondrial dysfunction, oxidative and proteostatic stresses, instability and impaired disposal of nucleoids cause the release of mitochondrial DNA (mtDNA) from the mitochondria in several human diseases, as well as in cell culture and animal models. Mitochondrial DNA mislocalized to the cytosol and/or the extracellular compartments can trigger innate immune and inflammation responses by binding DNA-sensing receptors (DSRs). Here, we define the features that make mtDNA highly immunogenic and the mechanisms of its release from the mitochondria into the cytosol and the extracellular compartments. We describe the major DSRs that bind mtDNA such as cyclic guanosine-monophosphate-adenosine-monophosphate synthase (cGAS), Z-DNA-binding protein 1 (ZBP1), NOD-, LRR-, and PYD- domain-containing protein 3 receptor (NLRP3), absent in melanoma 2 (AIM2) and toll-like receptor 9 (TLR9), and their downstream signaling cascades. We summarize the key findings, novelties, and gaps of mislocalized mtDNA as a driving signal of immune responses in vascular, metabolic, kidney, lung, and neurodegenerative diseases, as well as viral and bacterial infections. Finally, we define common strategies to induce or inhibit mtDNA release and propose challenges to advance the field.
Keywords: Circulating cell-free DNA; DNA-sensing receptors; Inflammation; Innate immunity; Mitochondria; Mitochondrial DNA.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
Assessing Mitochondrial DNA Release into the Cytosol and Subsequent Activation of Innate Immune-related Pathways in Mammalian Cells.Curr Protoc. 2022 Feb;2(2):e372. doi: 10.1002/cpz1.372. Curr Protoc. 2022. PMID: 35175686 Free PMC article.
-
Mitochondrial DNA release and sensing in innate immune responses.Hum Mol Genet. 2024 May 22;33(R1):R80-R91. doi: 10.1093/hmg/ddae031. Hum Mol Genet. 2024. PMID: 38779772 Free PMC article. Review.
-
Mitochondria in innate immune signaling.Transl Res. 2018 Dec;202:52-68. doi: 10.1016/j.trsl.2018.07.014. Epub 2018 Aug 7. Transl Res. 2018. PMID: 30165038 Free PMC article. Review.
-
Emerging views of mitophagy in immunity and autoimmune diseases.Autophagy. 2020 Jan;16(1):3-17. doi: 10.1080/15548627.2019.1603547. Epub 2019 Apr 21. Autophagy. 2020. PMID: 30951392 Free PMC article.
-
Mitochondrial DNA, oxidants, and innate immunity.Free Radic Biol Med. 2020 May 20;152:455-461. doi: 10.1016/j.freeradbiomed.2020.01.013. Epub 2020 Jan 17. Free Radic Biol Med. 2020. PMID: 31958498 Review.
Cited by
-
Hypoxic Status in COPD and ARDS Patients: Impact on Lipid Signature.Int J Mol Sci. 2025 Jul 3;26(13):6405. doi: 10.3390/ijms26136405. Int J Mol Sci. 2025. PMID: 40650185 Free PMC article.
-
New insights into interstitial cystitis/bladder pain syndrome at single-cell resolution.BJUI Compass. 2025 Aug 4;6(8):e70051. doi: 10.1002/bco2.70051. eCollection 2025 Aug. BJUI Compass. 2025. PMID: 40765664 Free PMC article. Review.
References
-
- Roger AJ, Muñoz-Gómez SA, Kamikawa R. The Origin and Diversification of Mitochondria. Curr Biol. 2017;27(21):R1177–92. - PubMed
-
- Lang BF, Burger G, O’Kelly CJ, Cedergren R, Golding GB, Lemieux C, et al. An ancestral mitochondrial DNA resembling a eubacterial genome in miniature. Nature. 1997;387(6632):493–7. - PubMed
-
- Pak O, Nolte A, Knoepp F, Giordano L, Pecina P, Hüttemann M, et al. Mitochondrial oxygen sensing of acute hypoxia in specialized cells - Is there a unifying mechanism? Biochim Biophys Acta Bioenerg. 2022;1863(8):148911. - PubMed
-
- Löffler M, Fairbanks LD, Zameitat E, Marinaki AM, Simmonds HA. Pyrimidine pathways in health and disease. Trends Mol Med. 2005;11(9):430–7. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
