

LSD1 inhibition attenuates targeted therapy-induced lineage plasticity in BRAF mutant colorectal cancer
- PMID: 40264166
- PMCID: PMC12016338
- DOI: 10.1186/s12943-025-02311-z
LSD1 inhibition attenuates targeted therapy-induced lineage plasticity in BRAF mutant colorectal cancer
Abstract
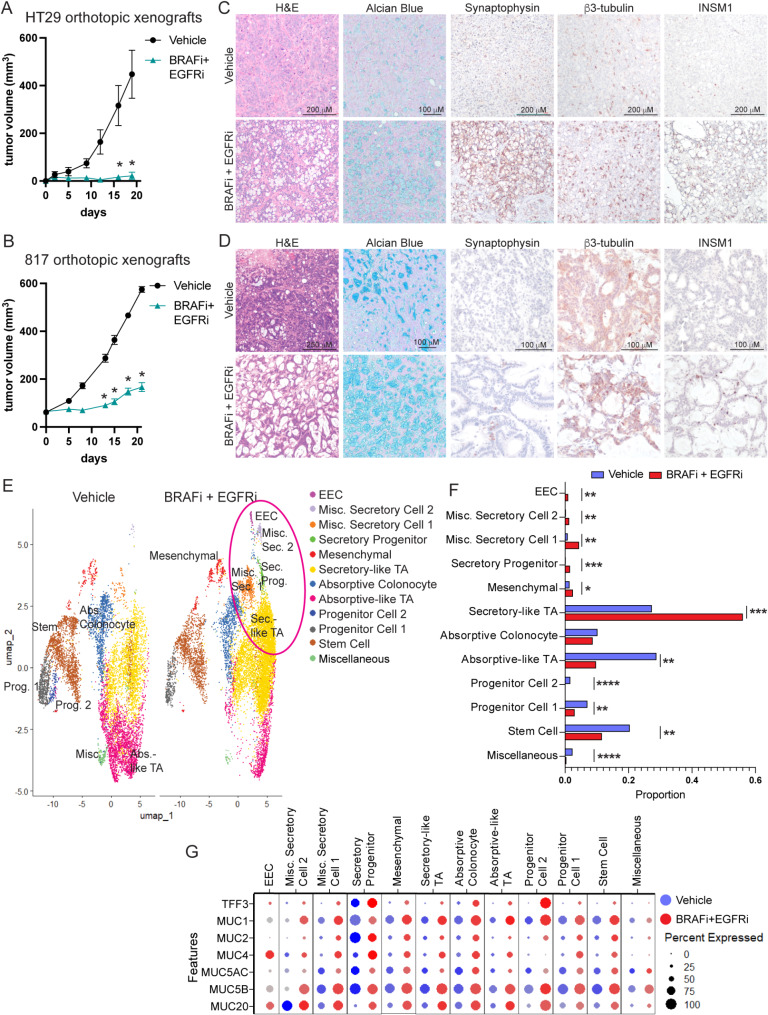
Background: BRAF activating mutations occur in approximately 10% of metastatic colorectal cancer (CRCs) and are associated with worse prognosis in part due to an inferior response to standard chemotherapy. Standard of care for patients with refractory metastatic BRAFV600E CRC is treatment with BRAF and EGFR inhibitors and recent FDA approval was given to use these inhibitors in combination with chemotherapy for patients with treatment naïve metastatic BRAFV600E CRC. Lineage plasticity to neuroendocrine cancer is an emerging mechanism of targeted therapy resistance in several cancer types. Enteroendocrine cells (EECs), the neuroendocrine cell of the intestine, are uniquely present in BRAF mutant CRC as compared to BRAF wildtype CRC.
Methods: BRAF plus EGFR inhibitor treatment induced changes in cell composition were determined by gene expression, imaging and single cell approaches in multiple models of BRAF mutant CRC. Furthermore, multiple clinically relevant inhibitors of the lysine demethylase LSD1 were tested to determine which inhibitor blocked the changes in cell composition.
Results: Combined BRAF and EGFR inhibition enriched for EECs in all BRAF mutant CRC models tested. Additionally, EECs and other secretory cell types were enriched in a subset of BRAFV600E CRC patient samples following targeted therapy. Importantly, inhibition of LSD1 with a clinically relevant inhibitor attenuated targeted therapy-induced EEC enrichment through blocking the interaction of LSD1, CoREST2 and STAT3.
Conclusions: Our findings that BRAF plus EGFR inhibition induces lineage plasticity in BRAFV600E CRC represents a new paradigm for how resistance to BRAF plus EGFR inhibition occurs. Additionally, our finding that LSD1 inhibition blocks lineage plasticity has the potential to improve responses to BRAF plus EGFR inhibitor therapy in patients.
Keywords: BRAF; Cetuximab; Colorectal cancer; Encorafenib; LSD1; Lineage plasticity; Neuroendocrine.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: All mouse experiments were covered under a protocol approved by the Indiana University Bloomington Animal Care and Use Committee in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care International. Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures




Update of
-
LSD1 inhibition attenuates targeted therapy-induced lineage plasticity in BRAF V600E colorectal cancer.bioRxiv [Preprint]. 2024 Oct 29:2024.10.25.620306. doi: 10.1101/2024.10.25.620306. bioRxiv. 2024. Update in: Mol Cancer. 2025 Apr 23;24(1):122. doi: 10.1186/s12943-025-02311-z. PMID: 39554172 Free PMC article. Updated. Preprint.
References
-
- Tabernero J, Grothey A, Van Cutsem E, Yaeger R, Wasan H, Yoshino T, et al. Encorafenib plus cetuximab as a new standard of care for previously treated BRAF V600E-Mutant metastatic colorectal cancer: updated survival results and subgroup analyses from the BEACON study. J Clin Oncol. 2021;39:273–84. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
