

Skeletal muscle effects of antisense oligonucleotides targeting glycogen synthase 1 in a mouse model of Pompe disease
- PMID: 40268518
- PMCID: PMC12017901
- DOI: 10.1002/ctm2.70314
Skeletal muscle effects of antisense oligonucleotides targeting glycogen synthase 1 in a mouse model of Pompe disease
Abstract
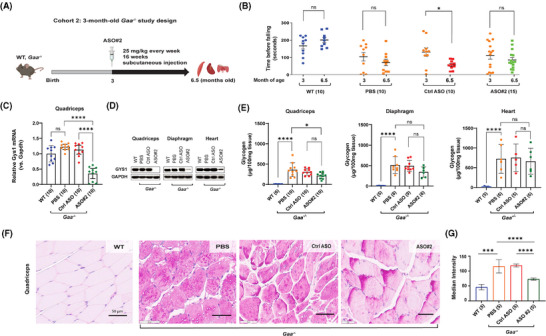
Pompe disease (PD) is a progressive myopathy caused by the aberrant accumulation of glycogen in skeletal and cardiac muscle resulting from the deficiency of the enzyme acid alpha-glucosidase (GAA). Administration of recombinant human GAA as enzyme replacement therapy (ERT) works well in alleviating the cardiac manifestations of PD but loses sustained benefit in ameliorating the skeletal muscle pathology. The limited efficacy of ERT in skeletal muscle is partially attributable to its inability to curb the accumulation of new glycogen produced by the muscle enzyme glycogen synthase 1 (GYS1). Substrate reduction therapies aimed at knocking down GYS1 expression represent a promising avenue to improve Pompe myopathy. However, finding specific inhibitors for GYS1 is challenging given the presence of the highly homologous GYS2 in the liver. Antisense oligonucleotides (ASOs) are chemically modified oligomers that hybridise to their complementary target RNA to induce their degradation with exquisite specificity. In the present study, we show that ASO-mediated Gys1 knockdown in the Gaa-/- mouse model of PD led to a robust reduction in glycogen accumulation in skeletal muscle. In addition, combining Gys1 ASO with ERT slightly further reduced glycogen content in muscle, eliminated autophagic buildup and lysosomal dysfunction, and improved motor function in Gaa-/- mice. Our results provide a strong foundation for validation of the use of Gys1 ASO, alone or in combination with ERT, as a therapy for PD. We propose that early administration of Gys1 ASO in combination with ERT may be the key to preventative treatment options in PD. KEY POINTS: Antisense oligonucleotide (ASO) treatment in a mouse model of Pompe disease achieves robust knockdown of glycogen synthase (GYS1). ASO treatment reduces glycogen content in skeletal muscle. Combination of ASO and enzyme replacement therapy (ERT) further improves motor performance compared to ASO alone in a mouse model of Pompe disease.
Keywords: Enzyme replacement therapy (ERT); Gaa‐/‐ mouse model; Pompe disease; antisense oligonucleotides (ASOs); glycogen synthase 1 (GYS1); skeletal muscle.
© 2025 The Author(s). Clinical and Translational Medicine published by John Wiley & Sons Australia, Ltd on behalf of Shanghai Institute of Clinical Bioinformatics.
Conflict of interest statement
Lan Weiss, Hong Yin, Pallabi Pal, Cheng Cheng, Lac Ta, Victoria Boock, Yasamin Fazeli, Mindy Chang, Marvin Paguio, Jonathan Lee, and Howard Yu report no disclosures. Nina Raben reports no disclosures. Virginia Kimonis is the Principal Investigator for the Rare Diseases Sanofi Registry and has received funding for an investigator initiated and outreach education programs for lysosomal storage diseases. Alyaa Shmara and Angela Martin have received fellowship funding from Sanofi‐Genzyme. Michele Carrer, Paymaan Jafar‐nejad, and Tamar Grossman are current or former paid employees of Ionis Pharmaceuticals.
Figures





Update of
-
Skeletal muscle effects of antisense oligonucleotides targeting glycogen synthase 1 in a mouse model of Pompe disease.bioRxiv [Preprint]. 2024 Mar 2:2024.02.22.580414. doi: 10.1101/2024.02.22.580414. bioRxiv. 2024. Update in: Clin Transl Med. 2025 Apr;15(4):e70314. doi: 10.1002/ctm2.70314. PMID: 38464319 Free PMC article. Updated. Preprint.
Similar articles
-
Skeletal muscle effects of antisense oligonucleotides targeting glycogen synthase 1 in a mouse model of Pompe disease.bioRxiv [Preprint]. 2024 Mar 2:2024.02.22.580414. doi: 10.1101/2024.02.22.580414. bioRxiv. 2024. Update in: Clin Transl Med. 2025 Apr;15(4):e70314. doi: 10.1002/ctm2.70314. PMID: 38464319 Free PMC article. Updated. Preprint.
-
Small molecule inhibition of glycogen synthase I reduces muscle glycogen content and improves biomarkers in a mouse model of Pompe disease.Am J Physiol Endocrinol Metab. 2024 Oct 1;327(4):E524-E532. doi: 10.1152/ajpendo.00175.2024. Epub 2024 Aug 22. Am J Physiol Endocrinol Metab. 2024. PMID: 39171753
-
A novel CD71 Centyrin:Gys1 siRNA conjugate reduces glycogen synthesis and glycogen levels in a mouse model of Pompe disease.Mol Ther. 2025 Jan 8;33(1):235-248. doi: 10.1016/j.ymthe.2024.11.033. Epub 2024 Nov 26. Mol Ther. 2025. PMID: 39604266
-
Gene Therapy for Pompe Disease: The Time is now.Hum Gene Ther. 2019 Oct;30(10):1245-1262. doi: 10.1089/hum.2019.109. Epub 2019 Sep 9. Hum Gene Ther. 2019. PMID: 31298581 Review.
-
Autophagy in skeletal muscle: implications for Pompe disease.Int J Clin Pharmacol Ther. 2009;47 Suppl 1(Suppl 1):S42-7. doi: 10.5414/cpp47042. Int J Clin Pharmacol Ther. 2009. PMID: 20040311 Free PMC article. Review.
References
-
- Raben N, Nichols RC, Boerkoel C, Plotz P. Genetic defects in patients with glycogenosis type II (acid maltase deficiency). Muscle Nerve Suppl. 1995;3:S70‐74. - PubMed
-
- Raben N, Nagaraju K, Lee E, et al. Targeted disruption of the acid alpha‐glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J Biol Chem. 1998;273:19086‐19092. - PubMed
MeSH terms
Substances
Grants and funding
- R21 AR080972/AR/NIAMS NIH HHS/United States
- R21AR080972-01/National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIH/NIAMS)
- The Council on Research, Computing, and Libraries (CORCL) at the University of California, Irvine
- The Helen Walker Award, Acid Maltase Deficiency Association (AMDA)
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous